Introduction
L-Glutamate is the major excitatory neurotransmitter in the central nervous system (CNS) and plays a dominant role in central excitatory neurotransmission involved in neuronal survival, synaptogenesis, neuronal plasticity, learning and memory processes. Glutamate is found naturally in millimolar levels in the brain; however, it can cause excitotoxic neuronal damage at high extracellular concentrations [1]. Extracellular glutamate concentrations are maintained within physiological levels by glutamate transporters (also known as excitatory amino acid transporters, EAATs) because no extracelluar enzymes exist for the breakdown of glutamate [2]. Five major subtypes of EAATs have been identified. EAAT1 is present in glial cells throughout the CNS and at high levels in the Bergmann glia of the cerebellum. EAAT2 is almost exclusively glial, and is widespread and highly abundant throughout the forebrain, cerebellum and spinal cord. The EAAT3 and EAAT4 are present predominantly in neurons; EAAT3 is selectively enriched in the neurons of the hippocampus, basal ganglia, cerebellum, and olfactory bulb, whereas EAAT4 is predominantly localized to cerebellar Purkinje cells, with low levels of expression also present in the forebrain. EAAT5 is present in the rod photoreceptor and bipolar cells of the retina [3,4].
Astroglial glutamate transporters (EAAT1 and EAAT2) are the predominant pathway for the synaptic inactivation of glutamate in the forebrain. Although there is little evidence for presynaptic or postsynaptic inactivation of glutamate by neuronal transporters, at least in the hippocampus and cerebral cortex, glutamate uptake could be primarily neuronal because many of the synapses are not surrounded by astrocytic processes in these brain areas [5]. Several studies have reported that EAAT3 activity increases with volatile anesthetics [6], propofol [7], lidocaine [8] and benzodiazepines [9], which could explain the anesthetic actions and the neuroprotective and antiepileptic effects of these drugs.
Clonidine, a selective α2-adrenergic receptor agonist, exhibits anesthetic, sedative, anxiolytic, sympatholytic and analgesic properties in clinical practice [10]. It has also been shown that clonidine is a potent neuroprotector in various cerebral hypoxia-ischemia models in which the accumulation of high concentrations of glutamate in the extracellular space is a key pathologic event and that α2-adrenergic receptors are important in mediating clonidine-induced neuroprotection [11,12]. It has been reported that activation of α2-adrenergic receptors located on the nerve terminals of the glutamatergic neurons inhibit the release of glutamate [13]. Considering these results, the sedative, anesthetic, and analgesic actions as well as the neuroprotective effect of clonidine may be implicated in the glutamatergic system, including the effect on the activity of the glutamate transporter. However, it is not known whether clonidine can affect the EAAT3 activity. The aim of this study is to investigate the effects of clonidine on the activity of EAAT3 proteins that can regulate extracellular glutamate using Xenopus oocytes.
Materials and Methods
Oocyte preparation and injection
Mature female Xenopus laevis frogs were purchased from Kato S Science (Chiba, Japan) and fed regular frog brittle twice weekly. All reagents, unless specified below, were obtained from Sigma (St. Louis, MO, USA). For removal of the oocytes, the frogs were anesthetized in 500 ml 0.2% 3-aminobenzoic acid ethyl ester in water until unresponsive to painful stimuli (toe pinching) and underwent surgery on ice. The oocytes were surgically retrieved and placed immediately in a calcium-free OR-2 solution. The OR-2 solution comprised (in mM): 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES with pH adjusted to 7.5. The oocytes were defolliculated with gentle shaking for approximately 2 hours in a calcium-free OR-2 solution including 0.1% collagenase type Ia. The oocytes were then incubated in a modified Barth's solution at 16℃ for one day before the injection of EAAT3 mRNA. The modified Barth's solution comprised (in mM): 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.41 CaCl2, 0.82 MgSO4, 0.3 Ca (NO3)2, 0.1 gentamicin and 15 HEPES, with pH adjusted to 7.5.
The rat EAAT3 complementary DNA (cDNA) construct was provided by Dr. Mattias A. Hediger (Brigham and Women's Hospital, Harvard Institutes of Medicine, Boston, MA, USA). The cDNA was subcloned in a commercial vector (BluescriptSKm). The plasmid DNA was linearized with a restriction enzyme (Not I) and mRNA was synthesized in vitro with a commercially available kit (Ambion, Austin, TX, USA). The resulting mRNA was quantified spectrophotometrically and diluted in sterile water. This mRNA was used for the cytoplasmic injection of oocytes in a concentration of 40 ng/ 40 nl by using an automated microinjector (Nanoliter 2000; World Precision Instruments, Sarasota, FL, USA). The oocytes were then incubated at 16℃ in a modified Barth's solution for three days before voltage-clamping experiments.
Electrophysiological recordings
Experiments were performed at room temperature (approximately 21-23℃). A single oocyte was placed in a recording chamber that was < 1 ml in volume and was perfused with 5 ml Tyrode's solution/min. The Tyrode's solution comprised (in mM): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgSO4, 10 dextrose and 10 HEPES, with pH adjusted to 7.5. Clamping microelectrodes were pulled from capillary glass (Single-Barrel Standard Borosilicate Glass Tubing, World Precision Instruments) on a micropipette puller (Temperature controlled pipette puller PIP5; HEKA Instruments Inc., Bellmore, New York, USA). The tips were broken at a diameter of approximately 10 µm and filled with 3 M KCl, obtaining a resistance of 1-3 MΩ. The oocytes were voltage-clamped using a two-electrode voltage clamp amplifier (OC725-A; Warner Corporation, New Haven, CT, USA) that was connected to a data acquisition and analysis system running on a personal computer. The acquisition system consisted of a DAS-8A/D conversion board (Keithley-Metrabyte, Taunton, MA, USA). Analyses were performed with pCLAMP7 software (Axon Instruments, Foster City, CA, USA). All measurements were performed at a holding potential of -70 mV. The oocytes that did not show a stable holding current of less than 1 µA were excluded from analysis. L-Glutamate was diluted in Tyrode's solution and superfused over the oocyte for 25 s (5 ml/min). The L-Glutamate-induced inward currents were sampled at 125 Hz for 1 min: 5 s of baseline, 25 s of L-glutamate application and 30 s of washing with Tyrode's solution. The glutamate-induced peak currents were calculated to reflect the amount of glutamate transported. We used 30 µM L-glutamate, unless indicated otherwise, in this study because the value of the pharmacokinetic parameter Km of EAAT3 for L-glutamate was shown to be 27-30 µM in previous studies [6,14].
Administration of experimental chemicals
Clonidine hydrochloride was dissolved in dimethyl sulfoxide and then diluted by Tyrode's solution to the appropriate final concentrations (0.2, 0.5, 2, 4, 7, 10, 20, 40 or 50 ng/ml that corresponds to 7.50 × 10-10, 1.88 × 10-9, 7.50 × 10-9, 1.50 × 10-8, 2.63 × 10-8, 3.75 × 10-8, 7.50 × 10-8, 1.50 × 10-7 or 1.88 × 10-7 M), which include clinically relevant plasma concentrations. In the control experiments, the oocytes were perfused with Tyrode's solution for 4 min before the application of Tyrode's solution containing L-glutamate for the electrophysiological recording. In the clonidine-treated group, the oocytes were perfused with Tyrode's solution for the first minute for stabilization followed by Tyrode's solution containing clonidine for 3 min before the application of Tyrode's solution containing L-glutamate for the electrophysiological recording. To determine the effects of clonidine on the Km and Vmax of EAAT3 and the reversibility of clonidine effects, the responses to serial concentrations of L-glutamate (3, 10, 30, 100, 300 and 1,000 µM) were assayed, the oocytes were then treated with 1.50 × 10-7 M clonidine and the responses were again recorded. For the stabilization, the oocytes were perfused with Tyrode's solution for 4 min before the second and third measurements.
Data analysis
Responses are reported as mean ± SEM. Each experimental condition was performed with the oocytes from at least three different frogs. Since the expression level of the transporter proteins in the oocytes of different batches may vary, variability in responses among the batches of oocytes is common. Thus, the responses were normalized to the mean value of the sameday controls for each batch. Similarly, in the reversibility experiments, the responses were normalized to the responses of the same oocytes to 1 mM glutamate under control conditions (before the clonidine treatment). This concentration of glutamate was the highest concentration used to induce EAAT3 activity and this normalization allowed us to pool together data from different batches of oocytes for analysis. Statistical analysis was performed using the Student's t-test, one-way repeated measures analysis of variance (ANOVA) or one way ANOVA followed by the Tukey multiple comparison test (SPSS version 18.0, SPSS Inc., an IBM Company, Chicago, IL, USA) as appropriate. Differences were considered significant at P < 0.05. Dose-response curves were prepared and IC50 was calculated using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA).
Results
The oocytes injected with EAAT3 mRNA showed inward currents after L-glutamate application (Fig. 1), whereas the uninjected oocytes were unresponsive to L-glutamate (data not shown). This current has been shown to be mediated via EAAT3 in previous studies [4,14].
In the vehicle control experiment, 0.04% (v/v) of dimethyl sulfoxide, the solvent for clonidine, (the highest concentration in Tyrode's solution containing clonidine) had no effect on the current response to glutamate (0.97 ± 0.11-fold) compared with the control (1.00 ± 0.13-fold) (n = 10, P > 0.05).
While clonidine itself did not induce any current in the oocytes injected with or without EAAT3 mRNA (data not shown), clonidine reduced the EAAT3 responses to L-glutamate (Fig. 1) in a concentration-dependent manner (7.50 × 10-10 - 1.88 × 10-7 M). The IC50 value (concentration required for 50% inhibition) of clonidine was 2.72 × 10-8 M. This inhibition was statistically significant at 2.63 × 10-8 - 1.88 × 10-7 M. Since the inhibition reached maximal at 1.50 × 10-7 M, we used this concentration for further experiments.
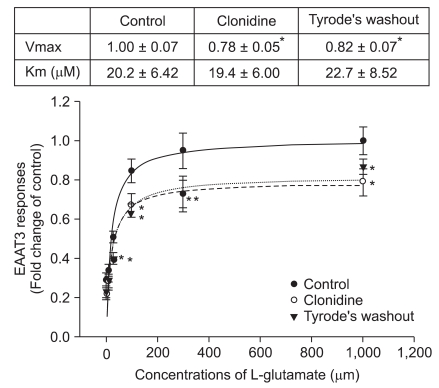
The Km of EAAT3 for L-glutamate was 20.2 ± 6.42 µM. This Km was not significantly affected by 1.50 × 10-7 M clonidine. However, clonidine significantly decreased the Vmax of EAAT3 for L-glutamate (from 1.00 ± 0.07 of control group to 0.78 ± 0.05 of clonidine group, P < 0.05). This clonidine effect decreased slightly, but not significantly in the oocytes perfused with Tyrode's solution for 4 min after clonidine treatment (Fig. 2).
Discussion
We demonstrated that clonidine did not have a significant effect on the activity of EAAT3, a major neuronal glutamate transporter, at clinically relevant concentrations (1.88 × 10-9 - 1.50 × 10-8 M, corresponding to 0.5-4 ng/ml), but that clonidine concentration-dependently reduced the EAAT3 activity significantly at higher concentrations (2.63 × 10-8 - 1.88 × 10-7 M, corresponding to 7-50 ng/ml).
EAATs are sodium co-transporters. They co-transport two or three sodium ions with one negatively charged glutamate molecule into the cell. Thus, at least one net positive charge enters the cell per glutamate transported. Thus, the transport of glutamate is electrogenic, and the size of the glutamate-induced current reflects the amount of glutamate transported. Therefore, measuring the glutamate-induced currents has been used widely in the literature to quantify EAAT activity [15]. Considerable evidence suggests that volatile, intravenous and local anesthetics may affect the EAAT3 activity [6-9,16].
Clonidine is an imidazoline derivate and a mixed α1 and α2 adrenoreceptor agonist with a predominant α2 action [17]. More recently, clonidine has been assuming greater importance as an anesthetic adjuvant because of its sedative, anxiolytic and analgesic properties in adult and pediatric practice [10]. The sedative/anesthetic-sparing properties of α2-adrenergic agonists are ascribable to the actions in the locus ceruleus, which is associated with a variety of physiologic regulatory processes, including regulation of sleep and wakefulness [17].
Arenas-López et al. [18] demonstrated a consistent and adequate level of sedation in the majority of cases at clonidine plasma concentrations of 0.9-2.5 ng/ml. Sumiya et al. [19] reported that the plasma clonidine concentration of 0.3-0.8 ng/ml would be sufficient to produce a satisfactory sedation in pediatric surgery. Clonidine is also known to be effective in the treatment of attention deficit-hyperactivity disorder at a therapeutic range of 0.5-4.5 ng/ml [20]. The exact clonidine concentrations around EAAT3 in the brains of patients on therapeutic doses of clonidine are not known. Clonidine crosses the blood brain barrier and the cerebrospinal fluid (CSF) concentration is 50% of the plasma concentration [21]. Thus, the therapeutic range of clonidine concentrations in the CSF may be lower than the clinically relevant plasma concentrations. Therefore, our results suggest that EAAT3 may not be a target for the sedative, anxiolytic, and anesthetic effects of clonidine.
We originally expected that clonidine may increase the EAAT3 activity because previous studies showed that volatile anesthetics, benzodiazepines, propofol and lidocaine increased the EAAT3 activity [6-9]. However, our results showed that clonidine rather reduced the EAAT3 activity significantly at 2.63 × 10-8 - 1.88 × 10-7 M (7-50 ng/ml). These concentrations are higher than the clinically relevant concentrations.
There is an apparent inconsistency in the literature concerning the proconvulsant, anticonvulsant and no effects of clonidine [22-24]. One of the working mechanisms of clonidine is the down-regulation of norepinephrine release form the locus ceruleus. It is known that the neurotransmitter norepinephrine plays a relevant role in modulating seizures [25]. Although interactions between central norepinephrine disturbances and seizure susceptibility are not fully understood, there is some evidence that norepinephrine may act as an anticonvulsant via the augmentation of the inhibitory γ-aminobutyric acid (GABA) effect [26]. The inhibition of neuronal EAATs induces epilepsy in rats and decreases the inhibitory post-synaptic current mediated by GABA in the rat hippocampus via a reduced synthesis of GABA because glutamate taken up by neuronal EAATs is a substrate for GABA synthesis [27]. Several studies on experimental animals indicated that the effects of clonidine change according to dose. Some studies have found an anticonvulsant effect of low-dose clonidine, whereas high doses decreased seizure thresholds [28,29]. Although a direct or indirect effect via another system(s) cannot be excluded, the glutamate accumulation due to a decreased EAAT3 activity may be suggested as one of the possibilities of the proconvulsant effect of clonidine.
We also demonstrated that clonidine did not affect the Km, but reduced the Vmax of EAAT3 for L-glutamate, suggesting that clonidine does not affect the affinity of EAAT3 for L-glutamate but decreases the total EAAT3 available for glutamate transporting. This regulatory mechanism of EAAT3 is consistent with the previous studies with regard to other drugs, such as volatile anesthetics [6], propofol [7], lidocaine [8], ethanol [14] and amitriptyline [30]. The clonidine-induced decreased EAAT3 activity was slightly, but not fully, recovered after a short washout, indicating that the clonidine effect may be irreversible. We could not clarify the exact reason for this in this study. It is believed that further study is needed with a longer duration of washout to determine the reversibility of clonidine effects. Possible limitations of this study are that the experiment was in vitro and that the mRNA of the rat instead of the human mRNA was used.
In conclusion, clonidine showed an insignificant effect on the activity of EAAT3 at clinically relevant concentrations, but decreased the EAAT3 activity significantly at higher concentrations. These results suggest that the inhibition of EAAT3 activity is not related to the sedation effect of clonidine. However, these results may provide additional data for the possible involvement of glutamatergic hyperactivity in the proconvulsant effect of clonidine.