Introduction
Reactive oxygen species (ROS) are normally generated at basal amounts, but ischemia or oxygen re-supply during reperfusion lead to abnormally high ROS levels that cannot be cleared by normal anti-oxidizing systems, causing tissue reperfusion injury [1]. An oxygen (O2) molecule generates the intermediate species, superoxide radical (O2·), hydrogen peroxide (H2O2), and hydroxyl radical (OH·), during the monovalent reduction reaction, and it is eventually converted to water (H2O) by catalase. In this process, the intermediates O2· and OH· are important free radicals that cause peroxidation (LOO·) of intracellular lipids and tissue damage [2]. In particular, OH·, the most reactive ROS [1], is produced by the Fenton reaction (Fe2+ + HOOH → Fe3+ + OH· + OH-) and the Haber-Weiss reaction (O2·- + NO· → OONO- + H+ → OONOH → NOO· + OH·)[3]. ROS generated during reperfusion causes organ damage in the heart [3,4], brain [5,6], and blood vessels [7,8].
In 1980, Furchgott and Zawadzki [9] found an endothelium-derived relaxing factor (EDRF) later shown to be nitric oxide (NO) [10]. Other vasoactive substances, including endothelium-derived hyperpolarizing factor (EDHF) and the endothelium-derived contracting factor (EDCF), are released from the endothelium to regulate vascular tone. However, because the endothelium is composed of a cell monolayer, it is easily damaged by physical and chemical stimuli, including ROS during reperfusion. Inhalation anesthetics [1,4,11] and lidocaine, a local anesthetics, can prevent ischemia-reperfusion injury in the brain [12].
In this study, we tested the effects of lidocaine and procaine, two local anesthetics, on endothelial-dependent relaxation in the rabbit aorta to examine if their anti-oxidizing effects could suppress or reduce the endothelial injury by ROS.
Materials and Methods
Preparation of ring slices and recording
All experiments conformed with the regulations of the Laboratory Animal Committee. Under enflurane inhalation anesthesia, ear intravenous injection of heparin 600 IU/kg was performed on rabbits, with exsanguinations 3 minutes later by cleaving the carotid artery. The abdominal aorta of the rabbits (2-2.5 kg, Male, n = 27) was extirpated and 3-4 mm long ring slices were prepared by separating the lipid tissue and connective tissue, not engaging any tension on them, in a petri dish with Krebs-Henseleit solution (K-H) solution (NaCl 120.0, NaHCO3 25.0, KCl 5.0, MgSO4 1.2, CaCl2 2.5, NaH2PO4 1.4, glucose 11.0 mM) bubbled with 95% oxygen and 5% carbon dioxide. Maintaining the temperature at 37 ± 0.5℃, one end of the aorta slice was fixed in the tissue bath with 5 ml of K-H solution. The opposite end was connected to the force displacement transducer (TSD 125C®, Biopac Inc., USA) and the K-H solution was exchanged every 15 minutes during the 90-minute equilibrating period. The final resting tension was fixed at 1.5 g. The vascular smooth muscle tension was recorded through an amplifier (DA100C®, Biopac Inc.) with a data acquisition system (MP100®, Biopac Inc.) and a personal computer.
Following the precontraction with phenylephrine (PE) 10-6 M, acetylcholine (ACh) 3 × 10-8, 10-7, 3 × 10-7, and 10-6 M were consecutively administered to observe the percent change (%) of the aortic tone. Changes of the aortic tone by ACh administration before ROS exposure (control) and after ROS exposure (experimental) were compared.
Method of ROS exposure
ACh-induced contractions were measured before and after electrolysis (EL) with an electric current (constant current, 15 mA) to the positive and negative electrodes in the K-H solution for 35 seconds to generate ROS and induce endothelial injury. More than 1 cm was maintained between the abdominal aorta ring slices and the positive and negative electrodes to avoid a direct stimulus of the electric field to the tissue. The K-H solution was then exchanged, followed by precontraction with PE and the second ACh treatment.
Vascular relaxation in the ROS scavenger pretreatment
To examine the action of the scavenger on ROS, 1,000 U/ml of catalase (n = 8), the hydrogen peroxide scavenger, was pretreated for 15 minutes. The hydroxyl radical scavengers, deferoxamine (n = 9) 0.1 mM, sodium salicylate 1 mM (n = 8), and mannitol 5 × 10-3 M (n = 12) were also pretreated, each for 30 minutes. Then, ROS was generated by EL to induce endothelial injury.
The lidocaine and procaine pretreatment groups
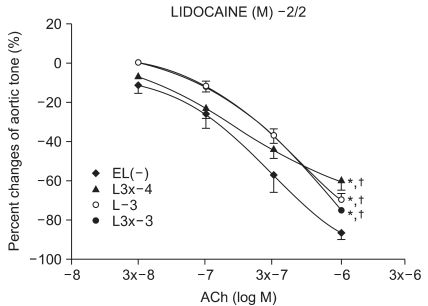
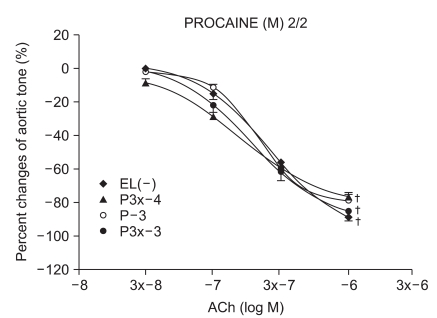
Experimental group values were obtained by 15 minutes of reagent pretreatment followed by ROS exposure. The experimental groups included pretreatment with lidocaine (L : n = 13, 13, 12, 10, 13, 11) or procaine (P : n = 6, 15, 12, 8, 10, 12) at concentrations of 10-5, 3 × 10-5, 10-4, 3 × 10-4, 10-3, and 3 × 10-3 M.
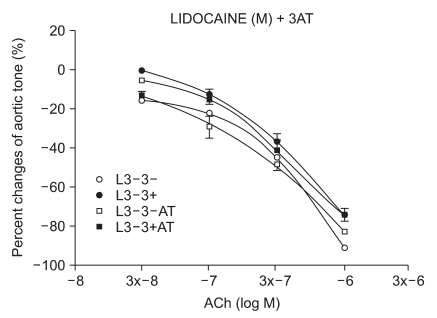
Effect of 3-amino-1,2,4-triazole (3AT) on endothelial injury by ROS
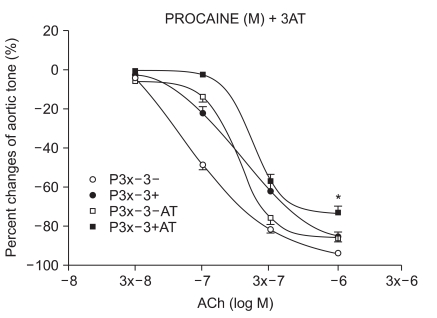
The samples were pretreated with 3AT, a catalase inhibitor, for 30 minutes. After a 15-minute waiting period, 3 × 10-4 M of lidocaine (n = 6) and procaine (n = 8) were pretreated for 15 minutes. Following 35 seconds of EL to generate ROS, ACh was added.
Reagents
All reagents, PE, ACh, lidocaine, procaine, catalase, defeoxamine, sodium salicylate, mannitol, and 3AT, were purchased from Sigma Co. (USA).
Statistical procedure
The degree of aortic tone relaxation by ACh was calculated as a percentage for control group values. Data are presented as mean ± SE. Prism 2.0® (GraphPad Software, San Diego, CA, USA) was used to obtain the dose-response curve by non-linear regression analysis. A paired t-test was used for the comparison of changes in each ring slice before and after ROS exposure, while an unpaired t-test was used for the comparison of the aortic tone after ROS exposure between the 3AT pretreated group and the non-pretreated group. For analysis of pretreated local anesthetic concentrations, a one-way ANOVA was used. All the post-hoc tests used the Dunnett test, with statistical significance as P < 0.05.
Results
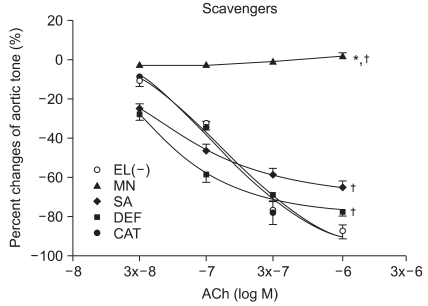
Vascular relaxation after ROS scavenger pretreatment
At ACh 10-6, the tension before ROS scavenger pretreatment was -87.6 ± 3.9%, and the endothelial-dependent relaxation was -87.6 ± 3.3, -77.6 ± 2.3, -65.6 ± 3.3, and 2.0 ± 1.8% with catalase, deferoxamine, sodium salicylate, and mannitol, respectively. The catalsase group was not affected by ROS, but the relaxation was significantly reduced in the other groups (P < 0.05, Fig. 1). The mannitol group did not show endothelial-dependent relaxation.
Discussion
We generated ROS by direct EL of a physiologic salt solution (PSS), such as the K-H solution [8], and the other researchers are directly injecting H2O2 to PSS [13]. ROS such as O2·, H2O2, and OH· are generated during PSS EL [14-17]. We did not add normal levels of ascorbic acid (1.1 × 10-4 M) to exclude its anti-oxidizing effect. A number of tiny air bubbles were generated at the positive and negative electrodes when the electric current was applied. In mouse thoracic aorta, 5 minutes of EL is sufficient to induce ROS injury [8]. However, 5 minutes of EL with 15 mA constant current caused too much injury in our model, so we used a 35-second treatment, indicating that there are species differences in the degree of injury by ROS.
Lecour et al. [18] verified that the function of the left ventricle declined when ROS was generated by 1 minute of EL in the K-H solution with a constant current of 1, 1.5, 3, 5, 7.5, and 10 mA, as measured by the Langendorff perfusion method. Superoxide radicals and hydroxyl radicals were also generated by the reaction of the 5, 5-dimethyl-1-pyrroline-N-oxide (DMPO)-OH signal, the adduct of DMPO EL, with superoxide dismutase (SOD : 100 IU/ml) and mannitol (50 mM), followed by a quantitative analysis with electron spin resonance. The DMPO-OH signal occurred as early as 10 seconds after starting EL and was maintained for 60 seconds, with diminishing levels after EL stopped. SOD pretreatement reduced the DMPO-OH signal by 60%, which showed that the hydroxyl radical generation process was partially by the Fenton reaction of a superoxide radical. Mannitol (50 mM) blocked the DMPO-OH signal but did not change cardiac parameters after EL [18]. Similarly, pretreatment of mannitol a scavenger of hydroxyl radicals, did not prevent endothelial dysfunction in our model.
ROS is also generated via cyclooxygenase during an inflammatory response [19]. Endotheliocytes, Kuffer cells, neutrophils, and macrophages also produce ROS and reactive nitrogen species (RNS) during an inflammatory response via antagonizing pathogenic bacteria in those cells. Antioxidant defenses are depleted by chronic exposure to ROS or RNS, leading to cancers, rheumatoid arthritis, or organ transplantation rejection. Large inflammatory responses also produce ROS, including peroxyl radical (ROO·), hydroxyl radical, superoxide radical, and hydrogen peroxide, and RNS, including ·NO and peroxynitrite anion (ONOO-).
Vascular relaxation induced by ACh is caused by inducing NO release in endotheliocytes, and ROS inactivates NO [20,21]. In addition, ROS generated by ischemia or reperfusion causes endothelial damage [15,22]. We generated ROS with 35 seconds of EL and then replaced the K-H solution for measurements after 15 minutes of equilibrium. Thus, ACh should induce vascular relaxation via changes in endothelial injury and not from inactivation of NO [20]. The endothelial monolayer is easily damaged and dysfunctional during chronic hypertension, even to the point of detachment [23]. Smooth muscle may also be directly injured by ROS.
During oxygen depletion, such as ischemia, ATP in the endothelium is rapidly consumed, causing failure of the Na+-K+ ATPase pump and an increase of intracellular [Na+] i and [H+]i by inhibiting the removal of the metabolites and mitochondrial NADH oxidation. Increased intracellular [H+] i leads to an increase in [Na+]i by facilitating Na+-H+ exchange to maintain constant [pH]i and thus triggers the Na+-Ca2+ exchange, increasing [Ca2+]i as well [24]. Increased [Ca2+] i deforms proteins and phospholipids [25] via activation of phospholipase A2 to generate arachidonic acid (AA), a free fatty acids. AA is then converted to prostanoids such as prostacyclin (PGI2), a strong smooth muscle relaxant, and prostaglandin G, a constrictor, to thromboxane A2 (TXA2) or to leukotrienes (LT), which cause the synechia of leukocytes to endotheliocytes. In addition, it facilitates the conversion of xanthine dehydrogenase to xanthine oxygenase (XO) and increases the intracellular superoxide radical (O2·) [23]. Oxygen re-supply after ischemia increases ROS: O2·, OH·, and LOO· [26]. These ROS that are generated mainly in mitochondria of neutrophil [25] denature lipids and proteins and damage the cell membrane and mitochondria (mitochondrial permeability transition pore, MPTP) [27], and denature enzymes [25]. O2· is the first ROS is generated by oxygen re-supply; it is produced by XO or by LT from neutrophils and nerve cells in the microvascular structure. Superoxide radical is converted by SOD to hydrogen peroxide, which eventually becomes water (H2O) by catalase. Here, catalase (1,000 U/ml) pretreatment prevented endothelial injury by ROS and maintained endothelial-dependent aorta relaxation. However, excess O2· is converted by the Fenton reaction and Haber-Weiss reaction to a hydroxyl radical, which causes the severe injury and eventually causes lipids peroxidation and cell and tissue injury by LOO· generation [26]. Pretreatment with mannitol (5 mM) as the OH· scavenger did not prevent endothelial injury, potentially because ROS sensitivity may shows species variation and higher doses may be needed.
Lidocaine protects against brain ischemia by reducing energy demand [28]. Anoxia-induced depolarization in the mouse hippocampus pretreated with lidocaine 1 µM and 0.25 µM 15 minutes before the ischemia-induction started after 49 seconds and 44 seconds, respectively, indicating a depolarization delay compared to the 27-second interval in the control group. The amount of ATP in the lidocaine pretreated group increased to 165% and 212% of controls at 1 minute and 2 minutes after ischemia, respectively. The activity of Na+-K+ ATPase and the activity of Ca2+ ATPase were not changed, and [Ca2+]i did not increase [28]. These findings imply that lidocaine maintains energy by delaying neuronal depolarization. In addition, [Ca2+] i increased in the left ventricle of the excised mouse heart where dysfunction was induced by hydrogen peroxide, but pretreatment with lidocaine 10 µM blocked this increase [29].
In human microvascular endothelial cells (HMVEC), lipopolysacharide in the culture solution caused necrosis of the endotheliocytes; amide local anesthetic pretreatment prevented this necrosis, but tetracaine and procaine, the esters, could not [30]. The protection by amide local anesthetics, including lidocaine, was blocked by 5-hydroxydecanoate (5HD), indicating that the mitochondrial ATP-sensitive K+ channel (mito[K+ATP]) is involved in the effect, consistent with the mechanism of ischemia-reperfusion injury. Because EL-induced injury from ROS involves direct injury by ROS that have not undergone the cascade reactions caused in the preconditioned cells, 5-HD pretreatment did not block the endothelial-protecting effect of lidocaine and procaine, indicating that mito[K+ATP] was not involved (data not shown).
We have tested whether ROS causes dysfunction in the rabbit aorta endothelium after lidocaine and procaine pretreatment. Lidocaine and procaine both dose-dependently protected endothelial-dependent relaxation by removing or anti-oxidizing ROS. Although lidocaine was less potent, both showed similar efficacy. In conclusion, pretreatment with lidocaine and procaine dose-dependently showed anti-oxidizing or ROS-removing effects to protect endothelial-dependent relaxation in the abdominal aorta after ROS exposure. Procaine could remove hydrogen peroxide as part of its protection mechanism.