Introduction
Certain amounts of reactive oxygen species (ROS) have been known to be generated in a normal body state, and can be also generated in large quantities by both ischemia or the oxygen resupply during ischemia reperfusion. Since ROS are not reduced by the normal anti-oxidizing system in a body, they can cause tissue reperfusion injury [1]. An oxygen (O2) molecule generates intermediate species, including: superoxide radical (O2┬Ę), hydrogen peroxide (H2O2) and hydroxyl radical (OH┬Ę) during the monovalent reduction reaction, and it is eventually converted to water (H2O) by catalase. In this process, the intermediates O2┬Ę and OH┬Ę are important free radicals that cause peroxidation (LOO┬Ę) of intracellular lipids and tissue damage [2]. The most reactive ROS, OH┬Ę, [1] is produced by the Fenton reaction (Fe2+ + HOOH ŌåÆ Fe3+ + OH┬Ę + OH-) and Haber-Weiss reaction (O2┬Ę- + NO┬Ę ŌåÆ OONO- + H+ ŌåÆ OONOH ŌåÆ NOO┬Ę + OH┬Ę) [3]. It has been revealed that the ROS generated during reperfusion causes organ damage in the heart [3,4], brain [5,6] and blood vessels [7,8].
After Furchgott and Zawadzki [9] revealed in 1980 that an endothelium-derived relaxing factor (EDRF) is released from vascular endothelium, Palmer et al. [10] proved in 1987 that the EDRF was nitric oxide (NO). Since then, it has been known that various vasoactive substances, including an endothelium-derived hyperpolarizing factor (EDHF) and an endothelium-derived contracting factor (EDCF), are discharged from vascular endothelium. Since such substances are discharged by various kinds of stimuli or a change in the conditions, vascular endothelium plays an important role in regulating vascular tone. However, because the vascular endothelium tissue is composed of a monolayer of cells, it is easily damaged by physical and chemical stimuli. Thus, the vascular endothelium tissue is the first target of ROS generated during reperfusion. Among the anesthetics currently used, inhalation anesthetics [1,4,11] and lidocaine, one of the local anesthetics, are known to be able to prevent ischemia-reperfusion injury in the brain [12]. In this study, we have evaluated the effect the COX (cyclooxygenase) blockers ketorolac and diclofenac, on rabbit aorta endothelium-dependent relaxation, to examine if their anti-oxidizing effect can suppress or reduce the vascular endothelium injury by ROS.
Materials and Methods
Preparation of ring slices and recording
All experiments were conducted conforming to the regulations of the Laboratory Animal Committee. Auricular intravenous injection of heparin 600 IU/kg was carried out in the rabbits under enflurane inhalation anesthesia and exsanguinations followed after 3 minutes by cleaving the carotid artery. The abdominal aortas of the rabbits (2-2.5 kg, Male, n = 27) were extirpated and 3-4 mm long ring slices were prepared by separating the lipid tissue and connective tissue, not engaging any tension on them, in a petri dish containing Krebs-Henseleit solution (K-H solution: NaCl 120.0, NaHCO3 25.0, KCl 5.0, MgSO4 1.2, CaCl2 2.5, NaH2PO4 1.4, glucose 11.0 mM) and 95% oxygen and 5% carbon dioxide were insufflated. While maintaining the temperature at 37 ┬▒ 0.5Ōäā, one end of the aorta slice was fixed in the tissue bath containing 5 ml of K-H solution, and 95% oxygen and 5% carbon dioxide was insufflated. The opposite end was connected to a force displacement transducer (TSD 125C┬«, Biopac Inc., USA) and the K-H solution was exchanged every 15 minutes during a 90 minutes equilibration period. The resting stage tension was fixed at 1.5 g. The vascular smooth muscle tension was recorded using an amplifier (DA100C┬«, Biopac Inc., USA) with a data acquisition system (MP100┬«, Biopac Inc., USA) and a personal computer.
Following the precontraction with phenylephrine (PE) 10-6 M, acetylcholine (ACh) 3 ├Ś 10-8, 10-7, 3 ├Ś 10-7 and 10-6 M were consecutively injected to observe the change of the aortic tone. Changes of the aortic tone by ACh injection before ROS exposure (control) and after ROS exposure (experimental) were compared.
Method of exposure to ROS
After obtaining the control group values by the consecutive injection of ACh, each reagent was pre-treated and, after a certain necessary interval, electrolysis (EL) was carried out by applying an electric current (constant current (CC), 15 mA) to the positive and negative electrodes in the K-H solution for 35 seconds to generate ROS and induce vascular endothelial injury. In the procedures, more than 1 cm of distance was maintained between the abdominal aorta ring slices and the positive and negative electrodes to avoid a direct stimulus of the electric field to the tissue. Then, the K-H solution was exchanged, the precontrcaction by PE followed, and ACh was consecutively injected. Changes of the aortic tone were recorded as the experimental group values. The electrode was made of platinum (7.5 mm) and 15mA of constant current generation was tested before the experiment.
Vascular relaxation in the ROS scavenger pretreatment
To examine the action of the scavenger on ROS, 1,000 U/ml of catalase (CAT, n = 8), the hydrogen peroxide scavenger, was pretreated for 15 minutes. The hydroxyl radical scavengers, deferoxamine (DEF, n = 9) 0.1 mM, sodium salicylate (SA) 1 mM (n = 8) and mannitol (MN) 5 ├Ś 10-3 M (n = 12) were also pretreated, each for 30 minutes. Then, ROS was generated by EL to induce vascular endothelium injury.
Effect of ketorolac and diclofenac pretreatment
After obtaining the control group values of the response to ACh before ROS exposure, the experimental group values were obtained by 15 minutes of experimental drug pretreatment followed by exposure to the ROS. The experimental groups included one group pretreated with ketorolac (K : n = 8, 10, 11, 9) and the other group pretreated with diclofenac (D : n = 7, 12, 13, 14), using concentrations of 10-5, 3 ├Ś 10-5, 10-4 and 3 ├Ś 10-4 M.
Effect of 3-amino-1,2,4-triazole (3AT) on vascular endothelial injury by ROS
The samples were pretreated with 3AT, the catalase inhibitor, for 30 minutes. After 15 minutes of waiting period, 3 ├Ś 10-4 M of ketorolac (n = 6) and diclofenac (n = 8) were additionally added and treatment was conducted for 15 minutes. Following 35 seconds of EL to generate ROS, ACh was consecutively injected.
Reagents
The reagents phenylephrine, acetylcholine, diclofenac, catalase, deferoxamine, sodium salicylate and mannitol, were purchased from Sigma Co. (USA). The ketorolac was Kerolmin® (Hana Pharmacy, Korea).
Statistical procedure
The percentages of ACh-induced relaxation of aortic rings were calculated and the results were used as the control group values. The data were presented as "mean ± standard deviation." Prism 2.0® (GraphPad software, USA) was used to obtain the dose-response curve by non-linear regression analysis. The paired t-test was used for the comparison of results before and after the ROS exposure, while an unpaired t-test was used for the comparison of the results of the 3AT pretreated group and the non-pretreated group. For the comparison of the results between the pretreated concentrations, a one-way ANOVA was used. All the post-hoc tests were carried out by the Dunnet test, defining statistical significance as P < 0.05.
Results
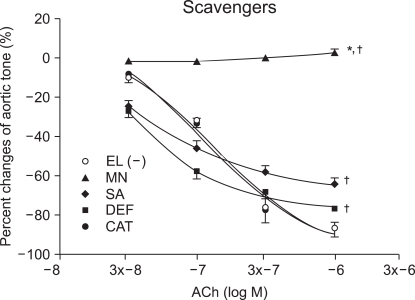
Vascular relaxation after ROS scavenger pretreatment
At ACh (10-6 M), the tension before ROS scavenger pretreatment was -87.6 ┬▒ 3.9% and the vascular endothelium-dependent relaxation was -87.6 ┬▒ 3.3, -77.6 ┬▒ 2.3, -65.6 ┬▒ 3.3 and 2.0 ┬▒ 1.8 % in the catalase, deferoxamine, sodium salicylate and mannitol, respectively. The catalase group was not affected by ROS, while the relaxation was significantly reduced in the other groups when compared with that of the catalase group (P < 0.05; Fig. 1). The mannitol group did not show vascular endothelium-dependent relaxation.
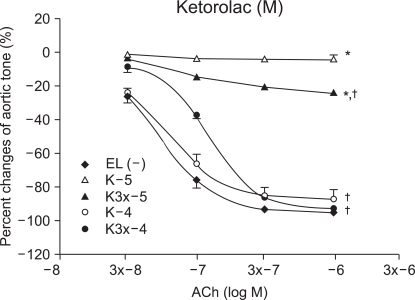
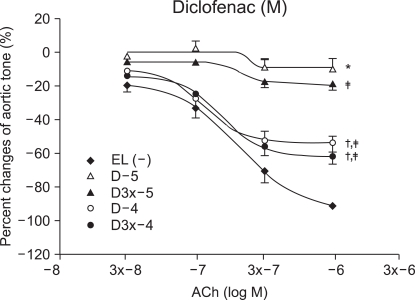
Vascular relaxation after vascular endothelial injury induction with ROS
The relaxation induced by ACh 10-7, (3 ├Ś 10-7 and 10-6 M groups) was significantly reduced by ketorolac (10-5 and 3 ├Ś 10-5 M) pretreatment, when compared with relaxation before the ROS exposure (P < 0.05; Fig. 2). The relaxation in the ketorolac pretreated groups (3 ├Ś 10-5, 10-4 and 3 ├Ś 10-4 M) was significantly increased after the ROS exposure, in proportion to the pretreatment concentration, when compared with the relaxation in the 10-5 M pretreated group (P < 0.05, 0.01, 0.01; Fig. 2). The relaxation of the diclofenac pretreated groups (10-5 and 3 ├Ś 10-5 M) was significantly decreased at ACh (10-7, 3 ├Ś 10-7 and 10-6 M), and the relaxation of the diclofenac pretreated groups(10-4 and 3 ├Ś 10-4 M) was significantly decreased at ACh (10-6 M) when compared with the relaxation before the ROS exposure (P < 0.05; Fig. 3). The relaxation of the diclofenac pretreated groups (10-4 and 3 ├Ś 10-4 M) was significantly decreased at ACh (10-7, 3 ├Ś 10-7 and 10-6 M), and the relaxation of the diclofenac pretreated groups (10-4 and 3 ├Ś 10-4 M) was significantly increased at ACh (10-7, 3 ├Ś 10-7 and 10-6 M), in proportion to the pretreatment concentration, after the ROS exposure, when compared with the relaxation of the diclofenac (10-5 M pretreated group) (P < 0.01, 0.01; Fig. 3).
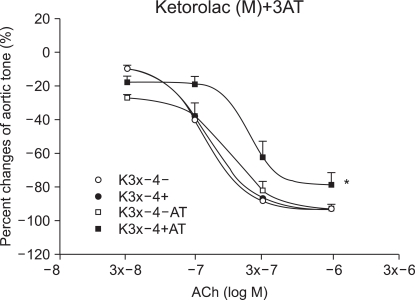
Effect of 3AT on the vascular endothelium injury by ROS
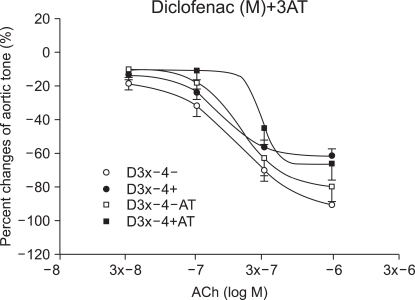
In the ketorolac pretreated group (3 ├Ś 10-4 M), which was pretreated with 3AT, the relaxation was significantly decreased at ACh (10-7, 3 ├Ś 10-7 and 10-6 M) (P < 0.01; Fig. 4 and 5). No significant change was found in the case of the diclofenac pretreated group (3 ├Ś 10-4 M) which was pretreated with 3AT (Fig. 6).
Discussion
The ROS experimental method includes the process to generate ROS by direct EL of physiologic salt solution (PSS), such as K-H solution, [8] and the process to directly inject H2O2 to PSS [13]. It has been known that ROS, such as O2┬Ę-, H2O2, OH┬Ę, are generated during the PSS EL [14-17]. Although ascorbic acid (1.1 ├Ś 10-4 M) is generally added to K-H solution it was not added in this experiment, to exclude its anti-oxidizing effect. We generated ROS by the EL method and fabricated the electrodes for this study. A number of tiny air bubbles were generated at the positive and negative electrodes when an electric current was applied. In one study, using mouse chest aorta, it was reported that 5 minutes of EL was required to induce injury by ROS [8]. However, 5 minutes of EL with 15 mA constant current caused too much injury by ROS in the rabbit abdominal aorta and the ROS generated by 35 seconds of EL was sufficient for our study. The implication of this experimental result is that there is an animal inter-specific variation in the degree of injury by ROS.
Lecour et al. [18] verified, in their study of the left ventricle function in a mouse heart using the Langendorff perfusion method, that the function of the left ventricle declined when ROS was generated by 1 minute of EL in K-H solution using a constant current of 1, 1.5, 3, 5, 7.5 and 10 mA. They also carried out EL in the cuvettes, each containing 125 mM 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), and quantitatively analyzed the signal of the adduct, DMPO-OH, using the electron spin resonance (ESR) method. In addition, they found that superoxide radicals and hydroxyl radicals were generated by the EL of K-H solution from the ESR experiments with the groups pretreated with superoxide dismutase (SOD : 100 IU/ml) and mannitol (50 mM), respectively. The DMPO-OH signal was found starting from 10 seconds after initiation of EL, stably generated until 60 seconds, when the EL was finished, and reduced afterward. In the case of SOD pretreatement, the DMPO-OH signal was reduced by 60%, which showed that the generation of hydroxyl radicals was partially by the Fenton reaction of a superoxide radical. On the contrary, the DMPO-OH signal was not found in the case of mannitol (50 mM) pretreatment. However, Lecour et al. reported that no significant difference in the cardiac parameter was found when the experiment was performed using the Langendorff perfusion method, in the group pre-treated with mannitol, a known hydroxyl radical scavenger, 15 minutes after the EL or the control group [18]. Similarly, in the scavenger pretreatment experiments in this study, pretreatment with mannitol could not prevent the vascular endothelium dysfunction by the ROS, in contrast to other scavengers' pretreatment.
The constant current applied for the quantitative analysis of the DMPO-OH signal by ESR was 1-40 mA, and the DMPO-OH signal corresponding to the amount of the generated hydroxyl radical was proportional to the magnitude of the constant current. In this experiment, 15 mA of a constant current was generated with a constant current amplifier to perform the EL for 35 seconds.
ROS is also generated as the result of cyclooxygenase (COX) action during inflammatory response [19]. Other major sources of ROS and reactive nitrogen species (RNS) formed during inflammatory response include vascular endothelium, Kuffer cells, neutrophils and macrophages. ROS and RNS are generated as the result of antagonizing infectious pathogenic bacteria in those cells. When ROS or RNS is continuously generated, or the anti-oxidizing defensive reaction is reduced, cancers, rheumatoid arthritis or organ transplantation rejection can result. The same diseases can be caused when the inflammatory response is extremely high, since the ROS, including peroxyl radicals (ROO┬Ę), OH┬Ę, O2┬Ę-, H2O2 and hypochlorous acid (HOCl), and RNS, including nitric oxide (┬ĘNO) and peroxynitrite anion (ONOO-), are increased. Thus, these are the pathophysiological findings of the above mentioned diseases, as well as the treatment targets. It has been known that indomethacin, the COX 1 and 2 blocker, can scavenge OH┬Ę, O2┬Ę-, NO┬Ę and ONOO- [19]. The half maximal inhibitory concentrations (IC50) for the hydroxyl radical scavenging activity of indomethacin, ketorolac and mannitol were 12 ┬▒ 2 ┬ĄM, 61 ┬▒ 7 ┬ĄM and 3,272 ┬▒ 215 ┬ĄM, respectively, indicating that the IC50 of mannitol was about 50 times higher than that of ketorolac [19].
It was reported that vascular relaxation induced by ACh is caused by induced release of NO in the vascular endothelium and that ROS inactivates NO [20,21]. In addition, the ROS generated by ischemia or reperfusion causes the vascular endothelium injury [15,22]. In this experiment, after ROS was generated by 35 seconds of EL, the K-H solution was replaced by a new solution and the measurement was carried out after 15 minutes of the equilibrium period. Since the measurement was done after the ROS was removed in this manner, blocking of the Ach vascular relaxation induction effect is assumed to be the result of the direct vascular endothelium injury by ROS, not the result of NO inactivation by ROS [20].
Because vascular endothelium tissue, a monolayer of cells, can be easily damaged, release of various vasoactive substances from vascular endothelium is impossible in the case of chronic hypertension, due to the damage, including the detachment of the vascular endothelium [23]. In this case, only smooth muscles exist in the blood vessel and they are directly damaged by the generated ROS. Thus, more studies are required regarding the damage of vascular smooth muscles.
In an oxygen depleted environment, such as ischemia or cardiac standstill, ATP in vascular endothelium is rapidly consumed, causing failure of the Na+-K+ATPase pump and an increase of intracellular [Na+]i and [H+]I by inhibiting removal of the metabolites and by mitochondrial NADH oxidation. Increased intracellular [H+]i leads to an increase of [Na+]i by facilitating the Na+-H+ exchange for the maintenance of constant [pH]I, thus triggering the Na+-Ca2+ exchange and increasing [Ca2+]i [24]. Increase of [Ca2+]i deforms the proteins and phospholipids [25]. In other words, increase of [Ca2+]i activates phospholipase A2 (PLA2), simulating generation of arachidonic acid (AA), one of the free fatty acids (FFA). The AA can then be metabolized and converted to prostanoids, such as prostacyclin (PGI2), which is a strong smooth muscle relaxant, prostaglandin G, which is a constrictor, thromboxane A2 (TXA2), or to leukotrienes (LT), which cause the adherence of leukocytes to vascular endothelium. In addition, it facilitates the conversion of xanthine dehydrogenase (XD) to xanthine oxygenase (XO) and increases the intracellular superoxide radical (O2┬Ę) [23]. Oxygen re-supply after ischemia increases 3 types of ROS: O2┬Ę, OH┬Ę and LOO┬Ę [26]. These ROS, which are generated mainly in the mitochondria of neutrophils [25], denature the lipid and protein and cause damage to intracellular structures, such as cell membranes and mitochondria (mitochondrial permeability transition pore, MPTP) [27], and can cause denaturation of enzymes [25]. O2┬Ę is the first ROS that is generated by oxygen re-supply. It is produced by XO or by LT from neutrophil and nerve cells in the microvascular structure. A superoxide radical is converted by superoxide dismutase (SOD) to hydrogen peroxide, which is eventually converted to water (H2O) by catalase. In this experiment, catalase pretreatment (1,000 U/ml) prevented vascular endothelium injury by ROS and a vascular endothelium-dependent aorta relaxation was expressed. However, it was reported that, when O2┬Ę- is generated in great quantities O2┬Ę- is converted by the Fenton reaction and Haber-Weiss reaction to a hydroxyl radical, which is the ROS that causes the severest injury and eventually causes lipid peroxidation plus cell and tissue injury by LOO┬Ę generation [26]. In our experiment, pretreatment with mannitol (5 mM), as the OH┬Ę scavenger, did not prevent vascular endothelium injury. Therefore, tests should be conducted using various mannitol doses.
We studied whether the induced rabbit aorta vascular endothelium relaxation caused by ROS is maintained following pretreatment with ketorolac or diclofenac. The result showed that vascular endothelium-dependent relaxation was maintained in proportion to the ketorolac and diclofenac pretreatment doses, verifying their ROS-removing or anti-oxidizing effect. The induced vascular endothelium-dependent relaxation by ROS was smaller in the diclofenac pretreated group when compared with that of the group treated with the same concentration of ketorolac. This implies that the IC50 for the anti-oxidixing action by diclofenac is larger and thus the ROS-scavenging capacity is smaller when compared with that of ketorolac. Hence, we expect that pretreatment with a high of concentration diclofenac will provide the same effect as that of ketorolac, but with an increased anti-oxidizing effect. On the other hand, Costa et al. [28] reported that non-steroidal anti-inflammatory drugs (NSAIDS) have a hydrogen peroxide scavenging effect. The H2O2-induced oxidation induced by lucinogen was measured using the chemilumiscence method and the resulting hydrogen peroxide scavenging activity potency ranking was as follows: sulindac sulfone > sulindac sulfide > GSH (reduced glutathione) > sulindac > indomethacin > acemetacin > etodolac > oxaprozin > ketorolac, melatonin > tolmetin. In our study, hydrogen peroxide experiments were conducted using catalase and 3AT, etc. The induced vascular endothelium relaxation was almost perfectly reproduced, even after the ROS exposure, following pretreatment with the H2O2 scavenger - catalase. Additionally, pretreatment with 3AT, a catalase inhibitor, resulted in a significant decrease of the induced vascular endothelium-dependent relaxation in the ketorolac group, showing that the hydrogen peroxide scavenging activity is partially involved in the anti-oxidizing activity of ketorolac.
In conclusion, it was shown that ketorolac and diclofenac have an anti-oxidizing effect on ROS or an ROS-removing effect. The hydrogen peroxide-removing effect was partially involved in the protective mechanism of ketorolac against ROS-induced vascular endothelial injury.