Intrathecal dexmedetomidine attenuates mechanical allodynia through the downregulation of brain-derived neurotrophic factor in a mild traumatic brain injury rat model
Article information

Abstract
Background
This study evaluated the effects of dexmedetomidine and propofol on brain-derived neurotrophic factor level in the cerebrospinal fluid (c-BDNF) and mechanical allodynia in a mild traumatic brain injury (TBI) rat model.
Methods
After fixing the rat’s skull on a stereotactic frame under general anesthesia, craniotomy was performed. After impact, 10 µl of drug was injected into the cisterna magna (group S: sham, group D: dexmedetomidine 5 μg/kg, group P: propofol 500 μg/kg, and group T: untreated TBI). The 50% mechanical withdrawal threshold (50% MWT) and c-BDNF level were measured on postoperative days (PODs) 1, 7, and 14.
Results
The 50% MWT measured on PODs 1, 7, and 14 was lower and the c-BDNF level on POD 1 was higher in group T than in group S. In group D, the c-BDNF level on POD 1 was lower than that in group T and was comparable with that in group S during the whole study period. The 50% MWT of group D was higher than that of group T throughout the postoperative period. In group P, there were no significant differences in the 50% MWT during the entire postoperative period compared with group T; the c-BDNF level was higher than that in group T on POD 1.
Conclusions
Intrathecal administration of dexmedetomidine may attenuate TBI-induced mechanical allodynia for up to two weeks post-injury through immediate suppression of c-BDNF in mild TBI rats. The inhibition of c-BDNF expression in the acute phase reduced the occurrence of TBI-induced chronic neuropathic pain.
Introduction
According to the World Health Organization, traumatic brain injury (TBI) resulting in death or disability affects approximately 10 million people annually [1]. TBI is preceded by sudden impact of an external force; as a result, the damaged brain tissue undergoes pathological changes, leading to dysfunction and neurological deficits [2]. The primary injuries include neuronal, axonal, glial, and vascular damage. The secondary injuries result from local biochemical responses and include excitotoxicity, inflammation, increased vascular permeability, and oxidative stress. The aberrant biochemical activities cause lesion expansion and neuronal cell death and worsen the dysfunction [3].
The majority (75–80%) of TBI cases is mild [4]. Mild TBI may be self-limiting and may often resolve completely without any neurological sequelae (e.g., motor deficits) [5]. Although mild TBI is associated with less severe clinical trauma, post-traumatic pain is more common in mild TBI than in moderate or severe TBI (75.3% vs. 32.1%) [6]. TBI-induced acute pain appears at the site of the primary injury and persists for the duration of its healing. The symptoms of pain and neurologic impairment may persist as chronic extra-cranial pain, post-traumatic headaches, sensory abnormalities, or mechanical allodynia [7]. Chronic pain may critically impact a patient’s quality of life, impeding daily recreational and occupational activities, with notable economic cost [8].
A systematic understanding of the underlying mechanisms and pathophysiology of TBI-induced pain is not yet established, and several theories have been proposed. A well-evidenced theory involves disruption of nociceptive signaling through induced dysfunction of descending inhibitory (e.g., noradrenergic) and ascending facilitatory (e.g., spinothalamic) pain pathways [9,10]. Furthermore, secondary injuries of neuronal cell death, neuroinflammation, excitotoxicity, neurodegeneration, and synaptic changes may contribute to chronic pain [7].
Brain-derived neurotrophic factor (BDNF), a ubiquitously expressed neurotrophin, and high-affinity receptor tropomyosin-receptor-kinase B (TrkB) are well-known for their roles in neuronal survival, maintenance, and plasticity [11,12]. BDNF has neuroprotective and neuro-regenerative functions in the central nervous system (CNS) and is closely related to the pathophysiology of several neurodegenerative diseases [12]. Based on the neuroprotective function of BDNF, its therapeutic potential in TBI has been assessed. However, infusions of BDNF in an experimental TBI model did not improve neuronal recovery or survival post-injury [13]. The BDNF-TrkB pathway has a documented association with neuropathic pain (NP) and hyperalgesia [11,14]. In descending pain pathways, BDNF plays a pro-nociceptive role in neuro-inflammatory and NP responses via the periaqueductal gray neurons within the rostroventral medulla and in the spinal cord [11,15]. Nociceptive sensitization corresponding with BDNF upregulation has been reported in rodent models of peripheral nerve ligation, spinal cord injury, and TBI [16–18]. TBI upregulates BDNF expression in the cortex, hippocampus, and spinal cord, inducing TBI-related pain sensitization [16,19]. It has been demonstrated that TBI-induced mechanical allodynia is correlated with elevations of BDNF level in the cerebrospinal fluid (CSF) (c-BDNF) [20].
Most patients with TBI admitted to the intensive care unit (ICU) require sedation [21]. Propofol and dexmedetomidine are the commonly used sedative agents in the ICU. In addition, propofol and dexmedetomidine are known to have properties that attenuate the occurrence of NP [22–24]. Dexmedetomidine and propofol, directly and indirectly, affect the BDNF-TrkB pathway [25,26]. This study hypothesized that dexmedetomidine and propofol would suppress the elevation of BDNF level after TBI, thereby alleviating the occurrence of TBI-induced mechanical allodynia.
This study aimed to investigate the effects of intrathecal (IT) administration of dexmedetomidine and propofol on changes in the level of c-BDNF and the incidence of mechanical allodynia caused by mild TBI.
Materials and Methods
Experimental animals, ethics statement, and randomization
All animal procedures were approved by the Institutional Animal Care and Use Committee of Pusan National University (approval number: PNUH-2019-157). All experiments followed the Guide for the Care and Use of Laboratory Animals published by the United States National Institute of Health (NIH Publication No. 85-23, updated 2011).
Seven-week-old male Sprague-Dawley rats weighing approximately 220 g were purchased from Hana Laboratories (Busan, Korea) and housed in a temperature-controlled room. Block randomization of 120 rats using an internet-based randomizer (https://www.sealedenvelope.com/simple-randomiser/v1/lists) allocated 30 age-matched rats each to one of the four groups as follows: an only craniotomy sham group (group S), an untreated TBI group (group T), a dexmedetomidine-treated TBI group (group D), and a propofol-treated TBI group (group P). The rats that expired were excluded from later analyses.
TBI modeling and treatment
Mild TBI was induced in the rat model by controlled cortical impact (CCI) [20] using an electromagnetically driven impactor device (PinPoint PCI3000 Precision Cortical Impactor, Hatteras Instruments, USA). The rats were briefly anesthetized (oxygen with 5% [vol/vol] and 2.5% [vol/vol] of isoflurane for induction and maintenance, respectively), and the skull was securely fixed to the stereotactic frame (U-frame stereotaxic instrument, Harvard apparatus, USA) while in the prone position. Craniotomy was performed using a mini drill with 5 mm trephine drill bits applied to the midline between the bregma and the lambda and 5 mm to the right of the sagittal suture (Fig. 1). The bone flap was then removed, and the tip of the CCI device was aligned vertically with the dura. The tip was subsequently positioned on the dura and delivered the impact with a depth of 3.5 mm, velocity of 3 m/s, and dwell time of 85 ms. We determined the set point of CCI based on the study of Schönfeld et al. [27]. After all processing was completed, the scalp was closed with a surgical stapler.
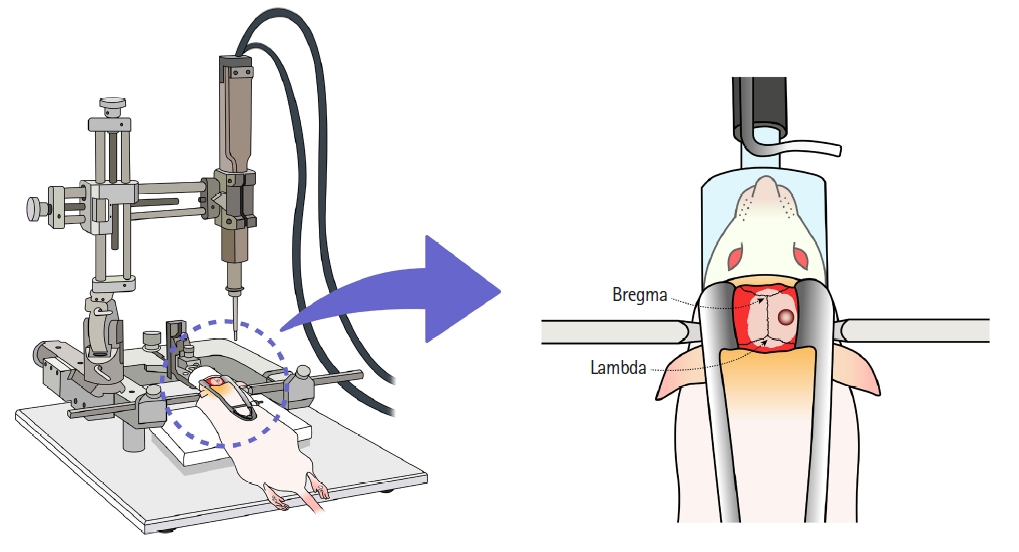
Illustration of rat head positioning for craniotomy during CCI. The skull of the anesthetized rat was securely fixed to the stereotactic frame in the prone position. Craniotomy was performed on the midline between the bregma and lambda and 5 mm to the right of the sagittal suture using a mini drill with 5-mm trephine drill bits. CCI: controlled cortical impact.
Immediately post-surgery, IT drug administration was performed under general anesthesia. The area of needle insertion was sterilized with betadine and alcohol. A 27 G disposable scalp vein set (Ningbo Greatcare Co., Ltd., China) was inserted vertically into the atlanto-occipital space. After confirming the free flow of CSF, 10 µl of drug (group D: dexmedetomidine 5 µg/kg and group P: propofol 500 µg/kg), pre-loaded in the scalp vein set, was slowly injected into the cisterna magna (Fig. 2A and 2B). At this time, if bloody CSF was visualized in the set or if CSF reflux was not observed, the procedure was immediately stopped and considered a failure.

Needle positioning for CSF collection and IT drug administration. (A) A 27 G scalp vein set connected with a 1 ml syringe was used for CSF collection and IT drug administration. After identifying the bony landmarks (triangle: external occipital protuberance, circle: atlas), the center of these two points was set as the needle entry point. (B) The needle was vertically inserted into the atlanto-occipital space. (C) The needle passes through the atlanto-occipital membrane and is placed in the cisterna magna. Once the needle is positioned correctly, clear CSF is pushed out into the tube attached to the needle. By the same method of needle insertion and using the same equipment, the drug was administered into the subarachnoid space. CSF: cerebrospinal fluid. IT: intrathecal.
Recovery time
Recovery time was measured to confirm the success of IT drug administration. The supply of inhalation anesthetic was removed, and the rats were placed in the supine position in the cage. Recovery time was defined as the time from anesthetic removal until the first change from the supine to prone position.
Pain behavior test
Before conducting the tests, the rats were habituated for 1 h on a metal mesh screen. The von Frey test for punctate mechanical allodynia was performed preoperatively and on postoperative days (PODs) 1, 7, and 14 by a blinded investigator to assess the 50% mechanical withdrawal threshold (50% MWT). A set of 11 von Frey filaments (vFF) was used (BIOSEB, In-Vivo Research Instruments, France) starting from vFF number 4.31. The vFFs were perpendicularly applied to the plantar surface of the left hind paws from underneath the screen, with sufficient force to bend the filaments. The vFFs were held in place for 2 s using the ‘up-down’ method [28]. The tests were continued until six additional stimuli after the first positive response had been recorded. If the response was continuously positive or negative, the stimulation was assigned a vFF number of 3.538 or 5.272, respectively.
The 50% MWT value was calculated using Dixon’s formula: 50% MWT = 10 (X+kd)/104; where X is the value of the final vFF number in logarithmic units, k is the tabular value of a positive/negative response, and d is the mean difference between the stimuli in logarithmic units (0.22) [29].
Mechanical allodynia was defined as a significant difference in the 50% MWT value comparing the experimental groups and group S at the same time point [16]. As an adjuvant method, mechanical allodynia was defined as a significant decrease in 50% MWT by comparing the preoperative and postoperative 50% MWT.
CSF sampling and storage
CSF samples were collected on PODs 1, 7, and 14. After anesthesia with isoflurane (oxygen with 5% [vol/vol] and 2.5% [vol/vol] of isoflurane for induction and maintenance, respectively), a 27 G scalp vein set (Ningbo Greatcare Co., Ltd., China) connected to a 1 ml syringe was vertically inserted into the atlanto-occipital space. A minimum CSF volume of 100 µl is required for BDNF enzyme-linked immunosorbent assay (ELISA) analysis. Therefore, 100 µl of CSF was collected by natural draining or gentle aspiration (Fig. 2C). On each experimental day, samples with volumes less than 100 µl or containing blood contamination were excluded from the analyses. Only grade 0 CSF samples (colorless and transparent [Fig. 3]) were analyzed. The samples were immediately stored in an icebox at 4°C. After centrifugation at 3000 rpm and 4°C for 15 min, the supernatants were collected and stored at −80°C.

Grade 0 CSF and CSF with blood contamination. CSF samples were classified into four grades according to blood contamination. Grade 0 is colorless and completely transparent. Grade 1 is slightly red but transparent. Grade 2 is slightly red and translucent. Grade 3 is red and opaque. By the same method of needle insertion and using the same equipment, the drug was introduced into the subarachnoid space. CSF: cerebrospinal fluid.
ELISA
c-BDNF levels were measured using ELISA (ProteinTech, USA), as per the manufacturer’s protocol. The BDNF in the samples was bound to the wells of a 96-well plate for 120 min using immobilized antibodies. Standard, blank, and CSF samples (100 µl each) were aliquoted in duplicate into the 96-well plate. After a buffer wash, a biotinylated anti-rat BDNF antibody was added, and the plate was incubated for 1 h. After a wash with buffer and addition of horseradish peroxidase-conjugated streptavidin, the wells were incubated for a further 40 min. The wells were washed with buffer, stained with 3,3′,5,5′-tetramethylbenzidine substrate solution for 20 min in the dark, and incubated with the stop solution. Absorbance was measured at 450 nm using a microplate-spectrophotometer (Synergy™ H1, BioTek Instruments Inc., USA) and converted to pg/ml, the selected unit for BDNF concentration. An average BDNF value was obtained from the duplicate samples.
Outcomes
The primary outcomes were changes in c-BDNF level and 50% MWT on PODs 1, 7, and 14 after mild TBI. The secondary outcomes were development of mechanical allodynia and mean weight change after mild TBI.
Statistical analyses
According to Cohen’s study, 18 subjects per group were required, with an alpha value of 0.05, a beta-value of 0.2, and a large effect size [30]. Estimating the dropout rate as 40%, 30 subjects per group were included. All analyses were performed using IBM SPSS Statistical Software (version 25; IBM Corp., USA) and MedCalc® Statistical Software (version 18.11.6; MedCalc Software Ltd., Belgium). The variables are presented as mean ± standard deviation or median and interquartile range. Normally distributed data were analyzed using ANOVA, and non-parametric data were analyzed using the Kruskal–Wallis test. If significant differences were reported, the Tukey-Kramer test and Conover test were performed for post-hoc multiple comparisons, as appropriate. Two-sided P values < 0.05 were considered statistically significant.
Results
Of the 120 rats, 30, 29, 30, and 30 rats were included in groups S, T, D, and P, respectively, for the final analyses (Fig. 4). The body weights and recovery times are presented in Table 1. The mean body weight of group P was significantly lower than that of group S on POD 14. The mean recovery time of groups D and P was significantly longer than that of group T on the day of IT drug administration (Table 1).
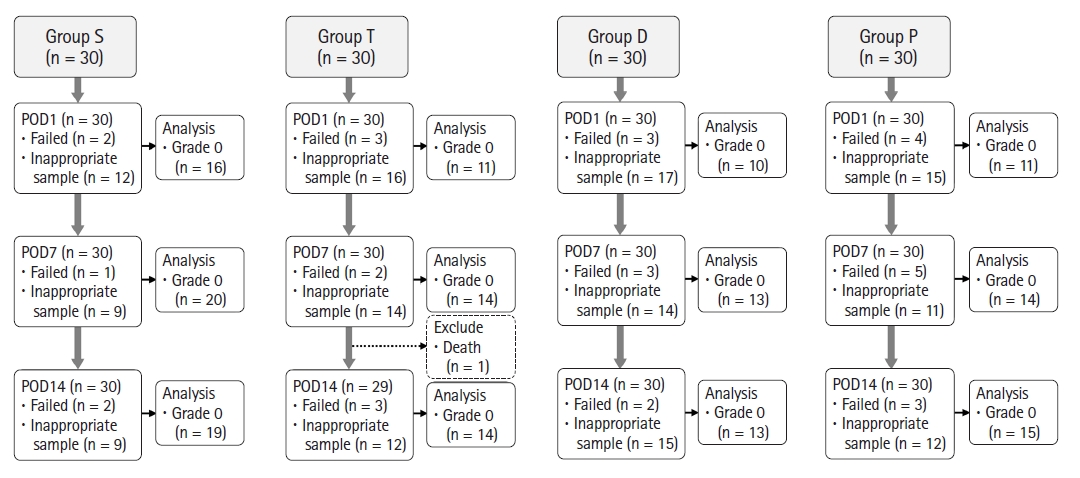
Flow chart of the selection process for the mild TBI rat model. For the quality control of sample analysis, samples less than 100 µl or not of grade 0 were considered as inappropriate samples. This figure shows the number of rats included in PODs 1, 7, and 14 for each group and the number of samples analyzed. All expired rats were excluded from later analyses. A minimum volume of 100 µl of grade 0 CSF was required for the analysis. All other samples were discarded. Group S: sham group, group T: untreated TBI, group D: dexmedetomidine treatment after TBI, group P: propofol treatment after TBI. TBI: traumatic brain injury, POD: postoperative day, CSF: cerebrospinal fluid.
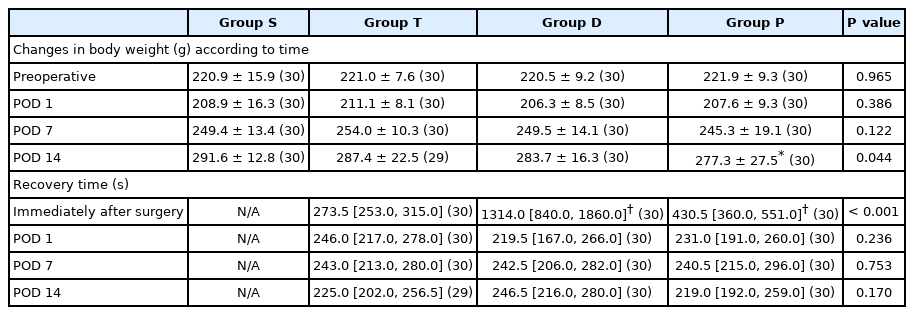
Changes in Body Weight according to Time and Recovery Time
The 50% MWT of groups T and P was significantly lower than that of group S during the entire postoperative period (Table 2). The 50% MWT of group D was significantly higher than that of group T on PODs 1, 7, and 14. There was no significant difference in the 50% MWT of group P compared with that of group T during the entire period. In group S, the baseline 50% MWT showed a lower trend on PODs 7 and 14, but there was no significant change during the entire period (Fig. 5). In group T, the baseline 50% MWT at PODs 1, 7, and 14 was 75.3% (47.8%, 94.9), 65.2% (32.0%, 94.9%), and 56.7% (42.1%, 105.1%), respectively, and showed significant differences compared with that at baseline (P < 0.001) (Fig. 5). In group P, the baseline 50% MWT at PODs 1, 7, and 14 was 84.5% (41.7%, 100.0%), 62.0% (45.5%, 100.0%), and 62.0% (41.7%, 90.4%), respectively, and showed significant differences compared with that at baseline (P < 0.001) (Fig. 5).
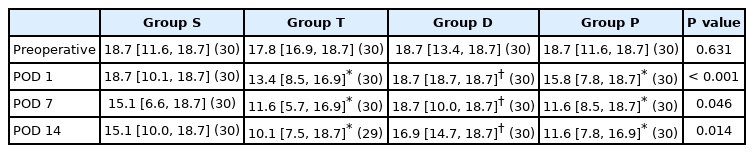
Change in 50% MWT (g) according to Time
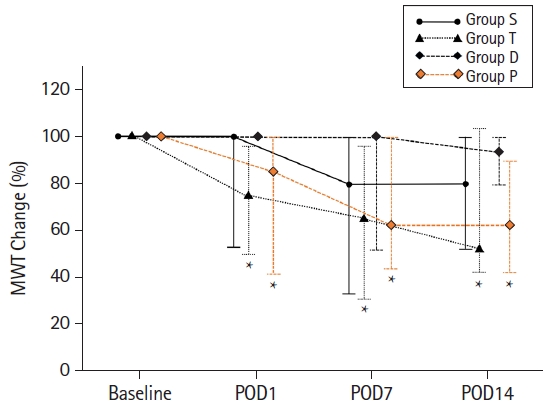
Changes in 50% MWT. The figure represents the change in the 50% MWT when comparing the 50% MWT of PODs 1, 7, and 14 with the preoperative value as the baseline. In groups T and P, the 50% MWT on PODs 1, 7, and 14 was significantly reduced compared with that at baseline. There were no significant changes in groups S and D. *P < 0.05 compared to the baseline. Group S: sham group, group T: untreated traumatic brain injury (TBI), group D: dexmedetomidine treatment after TBI, group P: propofol treatment after TBI. 50% MWT: 50% mechanical withdrawal threshold, POD: postoperative day.
On POD 1, group T showed significantly higher c-BDNF levels than group S (11.0 [10.0, 12.4] pg/ml vs. 8.6 [7.2, 9.8] pg/ml, P < 0.001) (Table 3). In group D, the c-BDNF level on POD 1 was significantly lower than that in group T (7.5 [4.7, 9.1] pg/ml vs. 11.0 [10.0, 12.4] pg/ml, P < 0.001) (Table 3). In group P, the c-BDNF level was higher than that in group T (14.3 [12.7, 17.0] pg/ml vs. 11.0 [10.0, 12.4] pg/ml, P < 0.001) on POD 1 (Table 3). On PODs 7 and 14, there was no significant difference in the c-BDNF level among all groups (Fig. 6).

Changes of BDNF (pg/ml) in CSF

BDNF level in the CSF. On POD 1, group T recorded significantly higher c-BDNF levels than group S. On POD 1, group D showed significantly lower c-BDNF levels than group T. In group P, c- BDNF levels on POD 1 were significantly higher than those in groups T and S. On PODs 7 and 14, there were no significant differences in c-BDNF levels among all groups. *P < 0.05 compared to group T. †P < 0.05 compared to the group S. Group S: sham group, group T: untreated traumatic brain injury (TBI), group D: dexmedetomidine treatment after TBI, group P: propofol treatment after TBI. c-BDNF: brain-derived neurotrophic factor level in the cerebrospinal fluid, POD: postoperative day.
Discussion
In this study, changes in the level of c-BDNF and the incidence of mechanical allodynia were observed after mild TBI. It was demonstrated that mild TBI induced an increase in the c-BDNF level and mechanical allodynia incidence. The administration of IT dexmedetomidine also suppressed the increase in the level of c-BDNF and the development of mechanical allodynia after mild TBI. However, IT propofol had no effect on the changes in the c-BDNF level and mechanical allodynia incidence after mild TBI.
Group T showed significantly higher c-BDNF levels than group S on POD 1. This increase in the c-BDNF level is thought to be a result of a neuroprotective and neuro-regenerative reaction due to the normal compensatory mechanism. Since this normal compensatory reaction did not occur in group D, the c-BDNF level was lower than that in group T. Due to this difference, group D may have an effect of inhibiting normal neuro-regeneration, but it is also thought to have an effect of inhibiting the generation of NPs. During a short-term follow-up, suppression of c-BDNF was associated with a decrease in the incidence of mechanical allodynia. However, further research is needed to determine what type of change would be observed at the long-term follow-up. In previous studies, the expression of BDNF was examined in CNS tissues [16,31,32]. When using CNS tissues, BDNF can only be measured at one time point; therefore, it was not possible to identify trends through continuous observation. Hence, this study was designed to measure the c-BDNF level, which was correlated with BDNF in the CNS and could be measured repeatedly [20]. In a human TBI study, the c-BDNF level was also significantly elevated at one day after TBI compared with that noted at baseline [32]. The results of this report were not different from our study results, which demonstrate that our model implemented mild TBI well [32,33].
A significant decrease in the 50% MWT was observed in groups T and P but not in group D. Although it was not statistically significant in group D, the 50% MWT was increased compared with that in group S. These results were consistent with the tendency shown in the difference in the c-BDNF level. That is, in groups D and S, wherein the c-BDNF level remained lower on POD 1, the 50% MWT was also maintained at a higher level than that in groups T and P. These results not only show that the expression of mechanical allodynia is related to the c-BDNF level but also imply that dexmedetomidine plays a role in suppressing the development of mechanical allodynia and that propofol does not play a role in suppressing it. The c-BDNF level in group P was significantly higher than that in group T; thus, it is questionable whether elevation of the c-BDNF level plays a role in promoting the development of allodynia. However, this will have to be elucidated through further experiments.
Dexmedetomidine and propofol are the commonly used sedative drugs in the ICU. Dexmedetomidine is a potent alpha-2 adrenergic agonist that is clinically applied for sedation and analgesia [34]. Although there is no known mechanism for the analgesic effect of dexmedetomidine, it has been found to be related to some downstream pathways of BDNF. Dexmedetomidine inhibits P2X4 receptor activation in microglia and induces the downregulation of p-38-mitogen-activated protein kinase and BDNF [35]. Propofol acts as an inhibitory neurotransmitter by binding to the gamma-aminobutyric acid (GABA) receptor [36]. Propofol induces the downregulation of protein kinase A-cAMP response element-binding protein-BDNF pathway [25,37]. GABA receptor inhibition, N-methyl-D-aspartate phosphorylation, and potassium-chloride cotransporter-2 protein blocker can activate the BDNF-TrkB pathway [11]. Therefore, dexmedetomidine and propofol were assumed to be related to the action of BDNF and the development of NP [26,38]. To the best of our knowledge, this study is the first to conduct IT dexmedetomidine and propofol administration and consequently follow-up the c-BDNF level, which may be related to the incidence of mechanical allodynia in a mild TBI rat model. We found that dexmedetomidine suppressed c-BDNF elevation but propofol did not. The results revealed in our study are partially consistent with those of previous studies but partially contradictory as well. This is presumed to be related to the action of drugs by another mechanism or the concentration of the drug or solvent.
As can be seen from our results, only the c-BDNF level measured on POD 1 showed a difference. However, for the 50% MWT of the contralateral hind paw, the difference in the results persisted not only on POD 1 but also on PODs 7 and 14. These results suggest that a transient increase in the BDNF level in the acute phase affects the occurrence of mechanical allodynia. Thus, the increase in the c-BDNF level in the acute phase is crucial for the development of mechanical allodynia. The mechanical allodynia in group T persisted up to POD 14, suggesting that the symptom was not self-limiting and might require intervention in the acute phase. In addition, after one significant increase, mechanical allodynia occurred even though the c-BDNF level was lowered, similar to that noted in other groups, on POD 7, suggesting that mechanical allodynia cannot be suppressed after it has already occurred. However, since the measurement was performed on POD 7 after POD 1, the interval was too long; more detailed observations are needed to determine whether there was a significant increase in the c-BDNF level from POD 1 to POD 7. In addition, since the timing of dexmedetomidine administration was immediately after the impact, further studies should examine whether a subsequent injection can also affect it. The immediate post-injury administration of dexmedetomidine resulted in early suppression of BDNF expression and resolution of pain symptoms in the acute phase. This was followed by suppression of mechanical allodynia development for two weeks thereafter. Dexmedetomidine effectively inhibited the development of chronic NP [22,35,39–41]. Therefore, the focus of therapeutic efforts should address a timely reduction in the c-BDNF level [42,43].
It was expected that normal weight gain would be interrupted by TBI-induced mechanical allodynia [44]. On POD 1, the body weight decreased in all the groups compared with that before brain injury. On POD 7, the body weight had returned to normal; the mean weight had increased post-operatively compared with that before brain injury. On PODs 1 and 7, postoperative differences in the mean body weight were not significant between the TBI groups (groups T, D, and P) and group S. In group P, weight recovery on POD 7 showed a tendency to be slow. On POD 14, there was a significant difference in the mean body weight of Group P compared with that in other groups. Craniotomy alone might affect normal weight gain. In addition, there has been a report that craniotomy itself can cause brain injury [45]. The causes of the weight difference are unknown due to a lack of research. Furthermore, propofol may inhibit or adversely affect weight recovery, and further studies are needed to verify this.
Recovery time was used for confirmation of IT drug administration. If dexmedetomidine and propofol were accurately injected into the cisterna magna, the recovery time would naturally extend. The recovery times of all subjects were prolonged on the day of IT drug administration. This confirmed that IT drug administration was performed properly in all the rats.
This study presented several challenges and limitations. First, a high dropout rate (52.6%) was recorded when considering all factors, resulting in exclusion; therefore, a large sample number is needed to analyze the results more reliably. IT catheterization is considered more appropriate for repeated CSF collection and IT drug administration. However, for several reasons, the IT catheter was not used in this experiment. There have been cases of the IT catheter detrimentally invading the spinal cord [46]. This indicates that IT catheterization itself can induce mechanical allodynia or affect the 50% MWT. IT catheterization is known to induce neuro-immune activation, resulting in an increase in glial markers and specific cytokines [47]. Thus, DeLeo et al. [47] did not recommend IT catheterization in neuro-immune-related studies. Therefore, we performed direct collection using a 27 G vein set instead of an IT catheter.
Second, definitive confirmation of NP in the nociceptive model is difficult to achieve. Late-onset NP after CNS injury is reported to occur even 1–3 years after the initial insult [48,49]. Thus, defining NP development in a TBI rat model is not realistically possible; therefore, mechanical allodynia was used as a reference for pain. Mechanical allodynia and NP after TBI are not the same; however, the results from the allodynia tests may extrapolate to NP and warrant further investigation. The standard 50% MWT for mechanical allodynia also varies across animals, and there is no known reference value. In comparison with the sham group, the 50% MWT that showed a significant difference was defined as mechanical allodynia [16].
Third, there is currently no evidence describing the mechanisms by which dexmedetomidine and propofol may alter the levels of c-BDNF and the effects thereof on pain responses. However, the study’s findings support a new avenue of investigation that may help better understand TBI-related pain pathophysiology and stimulate the development of novel therapeutic approaches to TBI treatment.
Fourth, we did not record observations on the presence or absence of rat hind paw paralysis. To evaluate NP using the vFF test, this study was designed to induce mild TBI that preserves the gross motor function. Schonfeld et al. [27] evaluated motor deficits in a rat model of CCI using an impact depth of 5 mm and velocity of 3 m/s and reported that, after CCI-induced TBI, rats showed impaired fine motor skills but intact gross motor skills. In our study, no rat exhibited paralysis of the hind paws or the fore paws.
In conclusion, the results of our study demonstrate that dexmedetomidine may attenuate TBI-induced mechanical allodynia for up to two weeks post-traumatic injury through immediate suppression of c-BDNF in a mild TBI rat model. The administration of IT dexmedetomidine was sufficient for correcting the mechanical allodynia over the measurement period. From a clinical perspective, management of the c-BDNF level in the acute phase may be a therapeutic goal for TBI-induced chronic pain.
Notes
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT) (No. 2018R1C1B6 004841).
This work was supported by clinical research grant from Pusan National University Hospital in 2022.
Conflicts of Interest
Eunsoo Kim has been an editor for the Korean Journal of Anesthesiology since 2016. However, he was not involved in any process of review for this article, including peer reviewer selection, evaluation, or decision-making. There were no other potential conflicts of interest relevant to this article.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author Contributions
Soeun Jeon (Data curation; Formal analysis; Investigation; Methodology; Resources; Software; Writing – original draft; Writing – review & editing)
Jiseok Baik (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Supervision; Writing – original draft; Writing – review & editing)
Jisu Kim (Visualization; Writing – original draft)
Jiyoon Lee (Investigation; Visualization; Writing – original draft)
Wangseok Do (Conceptualization; Data curation; Visualization; Writing – original draft)
Eunsoo Kim (Supervision; Visualization; Writing – original draft)
Hyeon Jeong Lee (Investigation; Supervision; Writing – review & editing)
Haekyu Kim (Conceptualization; Methodology; Supervision)