Perioperative implication of the endothelial glycocalyx
Article information

Abstract
The endothelial glycocalyx (EG) is a gel-like layer lining the luminal surface of healthy vascular endothelium. Recently, the EG has gained extensive interest as a crucial regulator of endothelial funtction, including vascular permeability, mechanotransduction, and the interaction between endothelial and circulating blood cells. The EG is degraded by various enzymes and reactive oxygen species upon pro-inflammatory stimulus. Ischemia-reperfusion injury, oxidative stress, hypervolemia, and systemic inflammatory response are responsible for perioperative EG degradation. Perioperative damage of the EG has also been demonstrated, especially in cardiac surgery. However, the protection of the EG and its association with perioperative morbidity needs to be elucidated in future studies. In this review, the present knowledge about EG and its perioperative implication is discussed from an anesthesiologist’s perspective.
Introduction
The luminal surface of the healthy vascular endothelium is covered with glycocalyx, a gel-like layer enriched with carbohydrates. Its dimensions vary according to the type of vasculature, ranging from 0.2 μm to more than 2 μm. The endothelial glycocalyx (EG) consists of various types of glycosaminoglycans covalently attached to plasma membrane-bound core proteoglycans. Heparan sulfate comprises 50–90% of endothelial glycosaminoglycans, and the remainder is a mixture of hyaluronic acid, dermatan, keratan, and chondroitin sulfates [1]. The high degree of sulfation of heparans, dermatans, keratans, and chondroitin sulfates, as well as the carboxyl groups of hyaluronic acid, contribute to the net negative charge of the EG, which influences the interaction between the EG and blood constituents. Two families of proteoglycans, syndecans 1–4 (single membrane-spanning domain) and glypicans 1–6 (glycosylphosphatidylinositol-anchored), provide the membrane-tethered scaffold for these glycosaminoglycans except hyaluronic acid, which is attached to the osteopontin receptor CD44. In addition, the EG incorporates diverse biologically active molecules, including extracellular superoxide dismutase, xanthine oxidoreductase, lipoprotein lipase, various cytokines, and regulators of coagulation. Several glycoprotein families (selectins, integrins, and immunoglobulins) are also incorporated in the EG. Pro-inflammatory stimuli increase the expression of these glycoproteins to act as adhesion molecules.
In a healthy vasculature, the EG and intercalated blood constituents form the endothelial surface layer (ESL). As a barrier between the endothelium and circulating blood, this layer regulates vascular permeability [2], as well as the interaction between endothelial and circulating blood cells [3,4]. The EG senses the shear stress of blood flow and transduces it into intracellular signals [5]. In addition, the binding and activation of various ligands and cell surface receptors may be affected by the EG [1,6]. The EG is not a static structure and is maintained in an equilibrium of continuous shear-induced shedding and synthesis. This constant state is, however, quite vulnerable and tends to disintegrate in response to various stressors, including ischemia-reperfusion, oxidative stress, hypervolemia, endotoxins, and other pro-inflammatory stimuli. The degradation of the EG leads to increased vascular permeability, facilitated transmigration of inflammatory cells, impaired mechanotransduction, reduced antioxidant activity, compromised anti-coagulant properties on the endothelial surface, and further activation and propagation of danger signals.
Physiologic Functions of the EG
Vascular permeability
In the classic Starling’s principle, the net fluid flux across the inter-endothelial junction is determined by the balance between hydrostatic and oncotic pressure gradients [7]. High hydrostatic pressure in the vascular lumen drives fluid to the interstitial space, whereas lower levels of proteins in the interstitial space compared to the plasma results in opposing forces for hydrostatic filtration. This can be expressed by the following equation:
where Jv/A is the filtration rate per area, Lp is the hydraulic conductance, Pc is the intravascular hydrostatic pressure, Pi is the interstitial hydrostatic pressure, σ is the reflection coefficient, πc is the plasma oncotic pressure, and πi is the interstitial oncotic pressure.
However, several quantitative studies revealed that actual fluid filtration differed from the amount predicted by the classic Starling’s principle. As hydrostatic pressure gradually decreases from the arteriolar to venular ends of the capillary, it was assumed that fluid is filtered to the interstitium along the arteriolar side of the capillaries and then reabsorbed at the venular side. This model has been refuted in later studies [8,9]. Revisions to Starling’s principle has been made, suggesting that the return of fluids to the circulation occurs exclusively via lymphatic drainage. In contrast to earlier experiments, which were conducted in models where the interstitial oncotic pressure was near zero [10], Levick [11] used accurate measures of interstitial oncotic pressure, which is almost half the plasma, and showed that the net filtration pressure predicted by the classic Starling’s equation was in far excess of the observed lymphatic flow. Moreover, when the interstitial oncotic pressure was raised, the increase in net fluid filtration was much smaller than expected [8]. These discrepancies indicated that interstitial protein concentration plays a minor role in the generation of the oncotic pressure gradient across the capillary wall. The introduction of the glycocalyx-cleft model helped to resolve the paradox, and Starling’s principle was revised to the following:
where πsg is the subglycocalyx oncotic pressure.
The EG acts as a molecular sieve of plasma proteins [9,12], and filtered fluid accelerates through the narrow junctional breaks. If this downstream fluid velocity approaches the upstream diffusion velocity of interstitial proteins, a washout of the diffusing proteins can maintain the protein concentrations in the sub-glycocalyx area at a very low level. Consequently, the effective oncotic pressure gradient opposing hydrostatic filtration is greater than the difference in oncotic pressure between the plasma and interstitium, reducing the basal fluid filtration rate to approximately the normal lymphatic drainage rate [13]. Enzymatic removal of EG components results in increased hydraulic conductivity, protein flux, albumin excretion, and edema [14–16]. When the negative charge of the EG was removed, increased permeability to albumin or dextrans were observed [17,18]. This evidence indicates that EG is a major determinant of vascular permeability.
In addition to a passive barrier role, recent studies suggested that the EG may regulate vascular permeability by acting as a mechanotransducer of flow-mediated shear stress [19]. According to Starling’s principal, hydrostatic pressure and fluid flux should have a linear relationship if Lp is held constant. However, it has been shown that increases in hydrostatic pressure results in an increase in Lp, thus resulting in a non-linear elevation of net fluid flux [20–23]. The shear-induced increase in Lp involves the activation of endothelial nitric oxide synthase (eNOS) and resultant alterations of junctional proteins [24]. The heparanase treatment of bovine endothelial cells abolished the shear-induced increase in Lp [23,25], suggesting a non-Starling mechanistic role of EG in the regulation of vascular permeability.
Mechanotransduction
One of the main roles of EG is represented by the mechanotransduction of blood flow-mediated shear stress to the cytoskeleton, nucleus, and enzymatic reactions, which is essential for vascular homeostasis. The EG functions as a sensor of mechanical forces exerted on the endothelial surface [26], in concert with other sensors including G-protein-coupled receptors [27], stretch-sensitive ion channels [28], rheological properties of the plasma membrane [29,30], caveolar structures [31], and integrins and focal adhesion molecules [32]. These mechanosensors initiate intracellular signaling such as increases in cytosolic calcium, the activation of eNOS [33], Rho-family guanosine triphosphatases, tyrosine kinases, Ras, Erk, and JNK; the phosphorylation of platelet endothelial cell adhesion molecule-1 (PECAM-1) intracellular domain [34]; and cytoskeletal remodeling [35], especially at the sites of highest tension like cell-cell junctions [36]. Physiologically, shear stress on the endothelium activates eNOS via the phosphorylation of S617, S635, S1177/S1179 residues by calmodulin kinase, PI3K/Akt, and PKA pathways [37]. In addition, shear stress also induces sirtuin 1, a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase, which enhances eNOS activity through deacetylation at Lys496 and Lys506 [38]. Physiological shear stress also induces transcription factors such as Kruppel-like factor-2 and nuclear factor-like-2, both of which are responsible for the induction of eNOS and antioxidant defense [39]. In contrast, in endothelial cells, perturbed blood flow induces the activation of the nuclear factor kappa B (NF-κB) pathway and activator protein-1, which participate in the acquisition of a pro-inflammatory phenotype by endothelial cells [40,41]. Defective mechanosensing and the resulting reduction in the shear-induced release of nitric oxide (NO) is associated with decreased eNOS activity. Direct pulling of the EG components and core proteins using atomic force microscopy results in a rapid Ca2+ influx and NO production [33,42], whereas enzymatic removal of EG [43], inhibition or knockdown of specific transient receptor potential (TRP) channels [44,45], or disruption of caveolae [46], all of which result in the impairment of flow-induced NO production.
NO has a crucial role in vascular homeostasis including the regulation of vascular tone, permeability, and inflammatory phenotype [47]. Decreased bioavailability and uncoupling of eNOS are associated with increased oxidative and nitrosative stress as well as endothelial dysfunction [48]. Of note, endothelial dysfunction induced by the disintegration of the EG and defective mechanotransduction may accelerate the further loss of the EG, leading to a vicious cycle of endothelial dysfunction. Decreased bioavailability of NO can increase exocytosis of lysosome-related organelles, which are responsible for the intrinsic mechanism of the EG degradation, via reduced S-nitrosylation of the N-ethylmaleimide-sensitive factor [49]. Furthermore, sirtuin 1 deficiency also affects vascular homeostasis. Recently, we reported that endothelium-specific sirtuin 1 knock-out mice have a significantly reduced whole body EG volume compared with wild-type mice [50]. Sirtuin 1-mediated deacetylation of Forkhead box O (FoxO1) DNA-binding protein is required for its active conformational change and enhanced cellular protection from oxidative stress [51,52]. Deficient sirtuin 1 expression or activity, which is associated with diverse vasculopathies [53], leads to susceptibility to oxidative stress. Oxidative stress activates disintegrin and metalloproteinase domain-containing protein 17 (ADAM-17) [54]. Activation of this enzyme contributes to cleavage of syndecan, which results in the shedding of EG [55]. Additionally, sirtuin 1 deficiency activates NF-κB, which induces transcriptional activation of heparanase, one of the target genes of this transcription factor [56,57]. Subsequently, increased heparanase activity leads to the enzymatic removal of heparan sulfate side chains from the EG and disintegration of ESL.
Role in blood cell-endothelial interactions
The EG shields the endothelium from interactions with circulating blood cells. The length of projections of cell adhesion molecules, including selectins (PECAM, vascular cell adhesion molecules (VCAMs), and ICAMs (intercellular adhesion molecules)) and integrins (CD11/CD18), are shorter than the thickness of the EG [58]. Thus, intact EG inhibits firm adhesion of leukocytes and platelets, whereas shedding of the EG facilitates their adhesion. In rodent cremaster muscle preparation, degradation of the EG by oxidized lipoproteins or tumor necrosis factor alpha (TNF-α) increased platelet or leukocyte-endothelial adhesion [3,59]. Similarly, chemotactic peptide fMLP (formyl-Met-Leu-Phe) degraded the EG and increased adhesion of leukocytes, while the matrix metalloprotease (MMP) inhibitor doxycycline attenuated the degradation of the EG and reversed the facilitated adhesion of leukocytes [4,60]. Knockout of syndecan-1 in mice resulted in increased adhesion and transmigration of leukocytes under pro-inflammatory stimuli as well as basal conditions [61].
Mechanisms of Glycocalyx Degradation
A variety of enzymes and reactive oxygen species (ROS) contribute to the degradation of the EG under inflammatory conditions. Activated neutrophils produce ROS and reactive nitrogen species (RNS), and release granules that contain proteases responsible for EG degradation [62]. Heparanase liberated from mast cells cleaves heparan sulfate side chains from membrane-bound core proteoglycans [63–65]. Additionally, hyaluronic acid can be cleaved by hyaluronidase. Although these enzymes are capable of fragmentation and removal of major glycosaminoglycans from the EG, more extensive shedding can occur by damage to core proteoglycans that constitute the backbone of ESL. Proteases released and activated under inflammatory conditions have been demonstrated to cause shedding of the EG [66]. MMPs are thought to cleave the syndecan ectodomain [60,66,67]. Other potential sheddases include neutrophil elastase, thrombin, plasmin, tryptase, and cathepsin B [68]. Upon appropriate inflammatory stimuli, phagocytes release their vesicles containing MMPs [67]. Doxycycline, a non-selective inhibitor of MMP activity, decreases shedding of the glycocalyx [60,66,67]. High affinity of MMP to heparan sulfate promotes the immobilization of MMPs within the ESL [69] and potentially enhances its destructive potential. Furthermore, MMPs have also been suggested to cleave CD44 [70–72].
ROS/RNS can damage the EG directly. One of the major sources of ROS is neutrophil-derived myeloperoxidase bound to the negatively charged glycosaminoglycan side chains [62]. The cleavage of HS after increased oxidative stress shows a similar pattern of increase in macromolecular passage compared to treatment with heparanase [73,74]. In addition, core proteoglycans are also susceptible to oxidative/nitrosative stress [62]. Furthermore, ROS/RNS can facilitate shedding of the EG via activation of MMPs and inhibition of endogenous protease inhibitors [54,62].
EG-degrading mediators are mainly released by inflammatory cells such as neutrophils and mast cells as described above. However, endothelial cells can be directly stimulated by inflammatory mediators such as TNF-α or lipopolysaccharides (LPS) to secrete molecules that contribute to the disintegration of the EG [59,75–78]. Recently, we showed using stochastic optical reconstruction microscopy (STORM) that the degradation of the EG is also mediated by the exocytosis of Weibel-Palade bodies (WPBs) and secretory lysosomes, which are visualized as patch loss or craters of the EG in a very early course of a rodent sepsis model [79]. Exocytosis of WPB and secretory lysosomes are one of the earliest responses of activated endothelial cells [80]. WPBs are rod-shaped organelles (0.2 μm × 2–3 μm) characteristic of endothelial cells and containing various kinds of proteins, enzymes, and inflammatory mediators. Known inducers of exocytosis of WPB include thrombin, histamine, leukotrienes, complements, superoxide anion, vascular endothelial growth factor (VEGF), sphingosine-1-phosphate, serotonin, vasopressin, and epinephrine [81–83]. The mechanisms of exocytosis of lysosomal-related organelles, such as WPB and secretory lysosomes, involves docking to the plasma membrane via soluble N-ethylmaleimide sensitive factor (NSF) attachment protein receptors (SNAREs) [81]. An interaction of NSF and its adaptor synaptosome-associated protein (α-SNAP) with SNARE proteins is required to prime WPB for exocytosis. This process can be inhibited by S-nitrosylation of NSF, implicating a crucial role of NO in the regulation of exocytosis of lysosomal-related organelles [49]. Notably, the prevention of exocytosis of lysosomal-related organelles with a NO donor improved survival of animals with severe experimental sepsis [79].
Perioperative Degradation of Glycocalyx
Acute degradation of the EG has been shown in patients with major surgery, especially those with cardiac surgery [84–88], as well as those with sepsis [89–92] and major trauma [93,94]. Elevated levels of EG degradation markers are associated with poor outcome in septic and critically ill patients [92,93,95–97]. In the perioperative setting, high postoperative syndecan-1 levels are correlated with greater incidence of severe acute kidney injury in pediatric patients that underwent cardiac surgery [98]. It has been proposed that ischemia-reperfusion (I/R) injury, oxidative stress, hypervolemia, and massive hemorrhage contribute to the perioperative degradation of the EG. I/R injury can disrupt the integrity of the EG through increased oxidative stress, secondary inflammatory response, and microvascular endothelial dysfunction [99,100]. The degradation of EG has been well demonstrated in animal models of mesenteric or cardiac ischemia [77]. I/R injury is also commonly encountered during cardiac, major vascular, and transplantation surgeries. Indeed, an elevation in the serum concentration of EG components, which reflects the degradation of the EG and release of its components into the circulation has been shown in cardiac and aortic surgeries [84–86]. Furthermore, I/R injury alone is not solely responsible for cardiac surgery-induced EG degradation. Cardiopulmonary bypass (CPB) induces an intense systemic inflammatory response, which is characterized by the activation of the coagulation system and complement pathway, I/R injury, recruitment of inflammatory cells in multiple organs, surge of pro-inflammatory cytokines, increased oxidative stress, activation of serine proteases, and microvascular endothelial dysfunction [101]. The inflammatory response following CPB is initiated by contact of blood with a non-endothelialized foreign surface and further accentuated by endotoxemia due to the translocation of LPS from the intestine to circulation [102–104]. Taken together, the inflammatory phenotype following CPB resembles that of sepsis, in which the degradation of the EG has been well-documented [105].
Interestingly, patients undergoing off-pump coronary artery bypass (OPCAB) showed a similar increase in the serum concentrations of the EG components, despite circumventing CPB [85]. This may have been due to the inevitable warm I/R injury from the temporary ligation of coronary arteries during grafting, hypotension, and the low cardiac output leading to the hypoperfusion of vital organs following cardiac displacement for the exposure of coronary arteries. Moreover, it was suggested that the release of atrial natriuretic peptide (ANP) during surgery may induce shedding of the EG. Cardiomyocytes in the atrium of the heart secrete ANP in response to a stretch in the atrium. In addition to its renal effect, ANP is known to increase microvascular permeability. Experimental studies demonstrated that ANP induced rapid shedding of EG via a cyclic guanosine monophosphate (cGMP)-linked proteolytic pathway, and resulted in increased net fluid flux and colloid extravasation to the interstitial space [106]. Furthermore, intracoronary infusion of ANP showed a dose-dependent effect on the EG shedding [107]. The displacement of the heart during OPCAB impairs ventricular filling and causes a stretch or compression of the atrium, leading to the release of ANP [108].
A reduction in the EG dimension was also shown in animal models of hemorrhagic shock [109,110]. However, the mechanism of shedding has not yet been elucidated. It is well known that patients with severe multiple trauma have diminished EG volume, probably due to massive hemorrhage and systemic inflammatory response [94]. Massive hemorrhage is not uncommon during major surgeries; however, no clinical study has addressed this issue so far.
Strategies to Protect EG
The natural regeneration of the EG appears to be slow. Animal studies suggest that the restoration of a hydrodynamically relevant volume of the EG following enzymatic degradation requires up to seven days [111]. There has been little evidence for the time-course of EG degradation and restoration during the perioperative period. In cardiac surgery, the concentrations of serum EG components peaked at the end of CPB, followed by a rapid decrease and return to near normal values at 24 h post-operation [84]. However, the serum concentrations of those fragments cannot be considered as a reliable indicator of EG regeneration, whereas an initial rise in serum can be a marker for degradation. Considering that the EG degrading stimuli can last several days postoperatively, the restoration of the EG may take a longer time. Therefore, strategies to minimize the degradation or accelerate restoration may have high clinical relevance.
Avoiding hypervolemia
Hypervolemia can result in the shedding of the EG, which is induced by the release of ANP due to atrial stretch. When colloid solutions are administered to normovolemic patients, 60% of the infused solution was extravasated immediately, whereas almost the entire volume was maintained within the intravascular space in an acute normovolemic hemodilution setting [112,113]. The prophylactic administration of fluids upon the induction of general or neuraxial anesthesia has long been advocated to counteract hypotension due to an anesthesia-induced decrease in cardiac preload. However, this practice cannot be recommended considering the role of the EG in fluid management. Furthermore, preloading of fluids did not show consistent efficacy for decreasing the incidence of hypotension or requirement for vasopressors after neuraxial anesthesia [114,115]. Moreover, liberal perioperative fluid administration leading to a positive fluid balance has been associated with increased morbidity [116]. Perioperative degradation of the EG may provide one rationale for restrictive fluid administration by a goal-directed protocol; however, the clinical impact of liberal versus restrictive fluids needs more extensive discussion [117] and is beyond the scope of this review.
Albumin
The EG can be stabilized by supplementation with albumin within the ESL [63]. The intercalation of albumin can provide protection against oxidative damage owing to an oxidizable sulfhydryl group of albumin, excessive attachment of leukocytes and platelets, and maintenance of shear-induced vasodilatation [63,118]. In addition, albumin carries sphingosine-1-phosphate, which inhibits MMP activity on the endothelium [119]. In an animal model for heart transplantations, the addition of histidine-tryptophan-ketoglutarate solution, which contains albumin reduced EG degradation [120]. In an experimental model of massive hemorrhage, resuscitation with plasma showed partial restoration of the EG, whereas lactated Ringer resuscitation did not [110]. These data suggest a possible role of albumin in the protection of the EG, although there is a paucity of direct clinical evidence.
Pharmacologic agents
A variety of pharmacological agents has been used to restore or prevent the degradation of the EG (Fig. 1). Agents that potentially interfere with EG degradation include antioxidants [121], antithrombin III [122], MMP inhibitor doxycycline [67], TNF-α analogue etanercept [123], NO donors [124], hydrocortisone [125], and volatile anesthetics [126,127]. However, these promising experimental study results have not yet been translated into clinical practice. Other methods to prevent EG degradation involve the supplementation of EG constituents such as heparan sulfate or hyaluronic acid [128]. Although accelerated restoration of the EG was achieved in vitro, heparan sulfate and hyaluronic acid could not reconstitute EG in vivo in a sepsis model [129]. The lack of effect in vivo can be explained by the overwhelming degradation process, which cannot be suppressed by the supplementation of EG components. Interestingly, we have reported a remarkable effect with sulodexide, an 8 : 2 mixture of fast-moving heparin and dermatan sulfate, on the restoration of the EG and survival benefit in a mouse model of severe sepsis [129]. The superior efficacy of sulodexide is thought to be due to its dual action, supplementation of EG constituents, and concomitant inhibition of degrading enzymes. Fast-moving heparin components of sulodexide, as a close mimic of heparan sulfate, are responsible for heparanase inhibition at basic clusters of heparanase in a competitive manner [130]. The dermatan sulfate fraction interacts with the active zinc binding site of the pro-MMP-9 molecule to block its conformational change into an active form [131]. It has also been reported that sulodexide attenuates the release of MMP-9 from leukocytes [132]. Clinically, Broekhuizen et al. [133] demonstrated that oral sulodexide administration for two months partially restored the thickness of the EG in patients with type 2 diabetes using sidestream dark field imaging of sublingual microcirculation. It is uncertain whether sulodexide would show a comparable effect in the perioperative setting. The required dose of sulodexide was estimated to be much higher to reconstitute the EG in a severe sepsis model [129]. Although sulodexide is known to exert minimal in vivo anti-coagulant effects due to its preferential uptake by the endothelium [134], care should be taken for its application in the perioperative setting because of a potential bleeding tendency from the administration of large doses. From this point of view, chemically modified, non-anticoagulant variants of heparin could be fascinating candidates for the protection of EG [135–137].
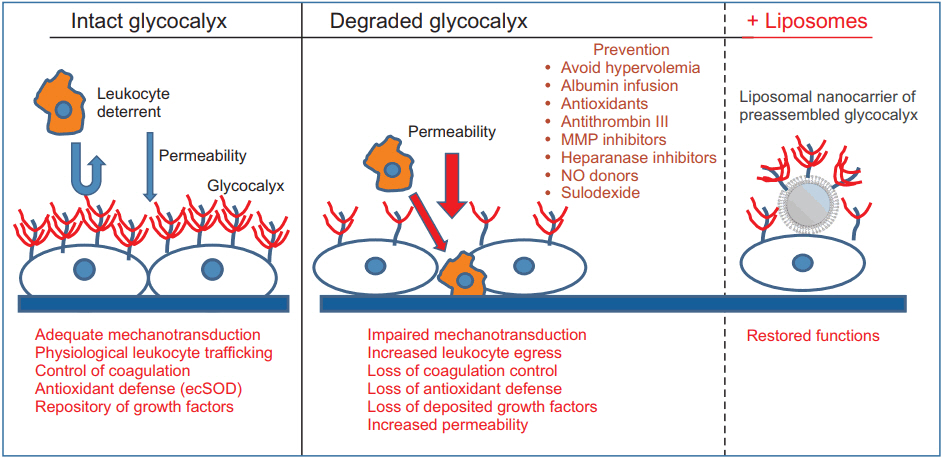
The physiological role of intact endothelial glycocalyx is represented. The degradation of glycocalyx leads to various pathologies. Possible tools for the restoration of glycocalyx are shown. MMP: matrix metalloprotease, ecSOD: extracellular superoxide dismutase, NO: nitric oxide.
Emerging Therapy to Restore the Degraded Glycocalyx
We have recently invented liposomal nanocarriers of the preassembled glycocalyx [138]. in vitro, ex vivo, and in vivo testing of these liposomes showed that they expeditiously restore glycocalyx in cultured endothelial cells stripped of EG by prior treatment with heparanase, restore mechanotransduction in isolated perfused arterioles, improve their flow-induced NO production, and partially restore renal microcirculation in LPS-injected mice. Collectively, these findings augur potential therapeutic benefits in restoring the EG in diverse conditions associated with the loss of EG.
Conclusion
The structure, function, and degradation of EG under various pathological conditions have been well recognized. I/R injuries, oxidative stress, hypervolemia, and systemic inflammatory responses promote EG degradation. Considering its unique location and diverse role in endothelial function, protecting the EG would probably improve clinical outcome after surgery. The degradation of EG during the perioperative period, especially in cardiac surgery, is a well-known phenomenon. Further study is needed to elucidate the clinical impact of EG degradation in the perioperative setting. Currently, no pharmacological tools for the restoration of EG are clinically available. However, attempts to minimize EG degradation, including avoidance of hypervolemia, and reducing the stress response and systemic inflammation, should be adopted by anesthesiologists. Furthermore, a novel strategy to restore degraded EG using liposomal nanocarriers of the preassembled glycocalyx is emerging.
Acknowledgements
Studies reported herein were supported in part by the grants from Westchester Community Trust.