Statistical and methodological considerations for reporting RCTs in medical literature
Article information

Abstract
Randomized controlled trials (RCTs) are known to provide the most reliable evidence on intervention. However, RCTs are often conducted and reported incompletely and inadequately, making readers and reviewers unable to judge the validity and reliability of the trials. In this article, we consider the statistical and methodological issues involved in reporting on RCTs, particularly in relation to the objectives, designs, and commencements of trials. This paper deals with the various issues that should be considered in presenting RCTs, and suggests checklists for reporting on them. We expect that these checklists will remind readers and reviewers to evaluate manuscripts systematically and comprehensively, making those manuscripts more transparent and reliable.
Introduction
What would happen if a researcher-intentionally or without knowledge-used a wrong research method or applied a right method in a wrong way and published these results? Or, what would happen if a researcher makes a wrong conclusion by misinterpreting or selectively reporting a methodologically correct study?
These reports would be valueless to physicians and patients, and would make it difficult for people to read and refer to the literature to assist their decisions in a number of areas, from patient care to national public health policies. Furthermore, it would waste limited resources-including time, money, and researchers' efforts-that could have been saved if the study had not been undertaken. A wrongly-conducted study also brings forth an ethical problem, in that the participating patients have been needlessly exposed to the risks involved in the study. Therefore, it is critical to plan, execute, and report studies appropriately [1,2].
Every study goes through a reviewing process to determine whether it should be published or not. There are three main types of reviews: professional peer reviews, which are conducted by experts in the field; ethical reviews, which are conducted by independent institutional review boards; and statistical reviews, which are concerned with statistics and epidemiologic methodology.
The main purpose of statistical reviews is to check that the study was carried out appropriately, that the results present integrity and accuracy, and that there is no error or selective reporting issue. Errors refer to all the elements that are not correct. There are two kinds of errors-systematic error and random error [2,3]. Random error occurs inconsistently, and is unrelated to the research process or the method. On the other hand, systematic error, which is another word for bias, occurs when the study is not correct, presenting problems in research process or method. The reviewers should take extra care to detect any systematic errors in carrying out statistical reviews [4,5]. It is often inevitable to introduce various errors into the research planning, commencement, and analysis process. It is therefore vital to minimize the risk of error in the research despite its inevitability.
Selective reporting, also called the "within-study publication bias," refers to cases in which the research results are not reported as originally planned. This also includes cases in which unplanned outcomes are added; or in which unplanned statistics and analyses of sub-categories or sub-groups are applied [6,7,8].
The study to assess the kinds and degrees of biases contained in the studies published on PubMed (http://www.ncbi.nlm.nih.gov/pubmed) was reviewed retrospectively, and conducted the follow-up survey to the authors of the studies. According to this study, 42 percent of the authors had not reported the efficacy outcomes completely, and 50 percent had not clearly specified the harm outcomes. The authors of this study argue that research protocols must be disclosed transparently in order to prevent medical studies from reporting selective and biased conclusions [9].
For this reason, established medical journals require peer reviewers for the randomized controlled studies to read the studies' research protocols prior to undertaking the actual reviewing process in order to confirm that the studies report all the objectives and outcomes according to their protocols.1), Journals such as PLOS One make it a requirement for authors to submit research protocols approved by Institutional Review Boards along with their papers.2), In its 'Instructions to Authors', The Korean Journal of Anesthesiology also recommends that authors register their clinical research protocol to protocol register site approved by WHO or International Committee of Medical Journal Editors.3)
In general, neither authors nor reviewers have the sufficient knowledge, training, and skills in statistical and methodological research issues. The problem is that a certain depth and range of knowledge about statistics and methodology is demanded of reviewers, and it is challenging to find a way to help reviewers meet this demand. Even experienced authors and reviewers often need guidelines in order to write or review research papers systematically and comprehensively with a consideration to statistics and methodology. For this reason, a number of journals provide checklists for authors and reviewers4,,5).
Guidelines provide practical information designed to help authors and reviewers report or review research methods and results by advising on the statistical and methodological elements that must be included in the research papers. By discussing these elements in this paper, we hope to provide a useful reference for authors and reviewers contributing to the Korean Journal of Anesthesiology. We also hope that our endeavor will help the papers published in the journal to meet statistical and methodological requirements, thus enhancing their quality and readability.
The main elements of statistical reviews include the study's objectives and design, the commencement of the trial, the methods of analysis, and the presentation and interpretation of the results. Errors in the analysis, presentation, and interpretation can be fixed by amending them. However, the paper will not be eligible for publication if it features errors in the objectives or design of the study, as these errors can only be eliminated by undertaking the study again from scratch. Therefore, it is essential to review statistical and methodological issues scrupulously at the early stages of setting the objectives and design. Hence, this paper first discusses the objectives, design, and progression of research (Table 1).
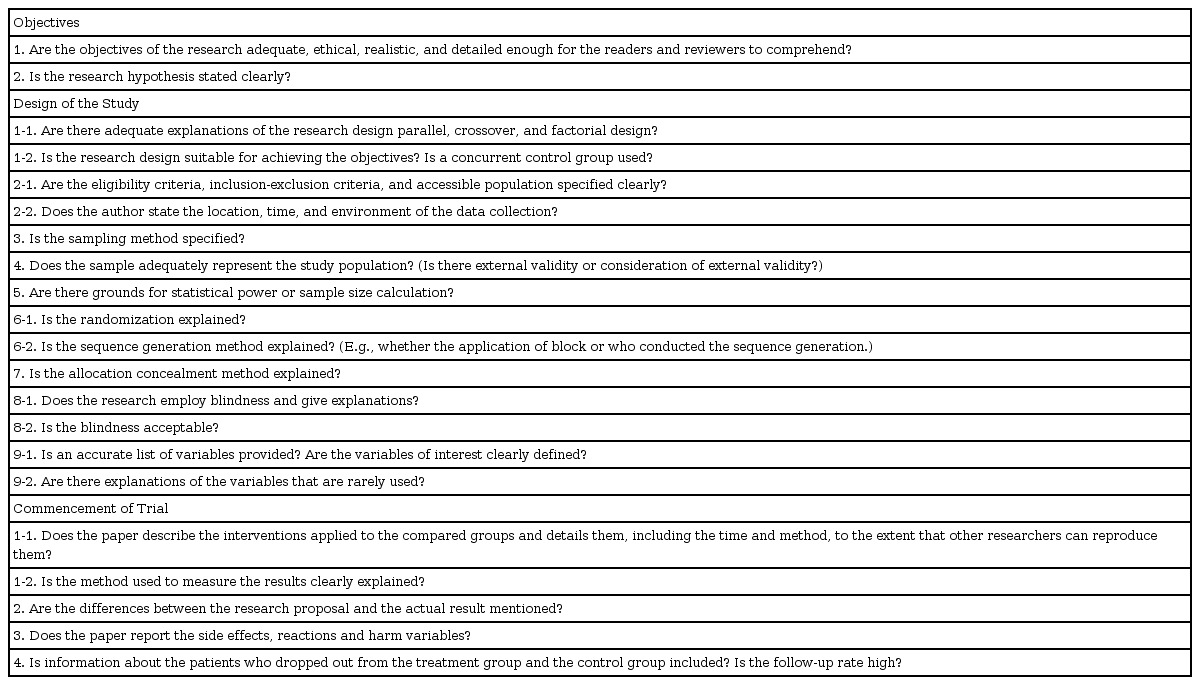
Checklist for Statistical and Methodological Considerations
Objectives and Hypothesis
1. Are the objectives of the research adequate, ethical, realistic, and detailed enough for the readers and reviewers to comprehend?
Research involves a series of processes, from the formulation and testing of research hypotheses to rebuttals and outcomes. Objectives are the desired answers to the research questions. In medical research, objectives are often linked to the curing or preventive effects of a certain medical intervention. Objectives should be valuable on their own. They should be ethical, and detailed enough for the readers and reviewers to understand. Moreover, they should not be overly broad or unrealizable by requiring too much time, cost, or other resources. Furthermore, not only the primary, but also the secondary objectives must be clearly stated in the research protocol in the planning stages. The results of the stated objectives need to be included in the research paper or dissertation.
2. Is the research hypothesis stated clearly?
A hypothesis is a question that has been formulated to help achieve the objectives of the research. Hypotheses are more specific and explicit than objectives, making them suitable for statistical testing. Good research starts with a good question based on a good hypothesis, and a good hypothesis should be based on the sufficient scrutiny of data from pre-clinical and clinical trials. Therefore, the quality of the research depends on the quality of the hypothesis. If the research hypothesis is unclear, or if there are multiple hypotheses to be tested, not only the scientific but also the statistical hypotheses must be clarified before initiating the research.
Research Design
1-1. Are there adequate explanations of the research design (parallel, cross-over and factorial design)?
1-2. Is the research design suitable for achieving the objectives? Does it use a concurrent control group?
Most problems in research spring not from wrong analyses, but from inadequate designs. Therefore, a good study design is vital to successful research [10]. In experiment research which should be distinguished from randomized controlled trials, there are studies which used historical control or non-randomized concurrent control. Control groups must be needed when assessing the effectiveness of an intervention. One possible way to do this is to use the past data of similar patients (historical control). This method may seem attractive to researchers on account of its simplicity and convenience, saving them from recruiting new study participants. In those cases, however, they have to use medical records for the historical control group, which may not be suitable for research as medical records are made for clinical purposes. These data may differ from the data collected for the research in terms of experimentation (or examination) conditions or research methodology. There may be a number of important unreported categories of data, and strict research criteria may not have been applied. Moreover, it may be difficult to determine whether the discrepancies among the groups of patients are due to the intervention or to other factors, since the environment, lifestyle, and adjuvant care differ according to differences in time. Logically, studies that use historical control groups cannot be altered to randomized control trials. However, because of confusion, some authors wrongly state that they are. Such logical errors make papers less persuasive to readers and reviewers. Non-randomized control trials may solve the problems of historical control groups, but may be influenced by confounding or biases, which will be discussed further down in this paper [11]. There are a few kinds of randomized control trials-including parallel design, cross-over design, and factorial design-, and every research should evidently state which design was used.
2-1. Are the eligibility criteria, accessible population, inclusion and exclusion criteria specified clearly?
2-2. Does the author state the location, time, and environment of the data collection?
The population is the entire set of subjects for the interests. If researches were to be conducted over the whole population, the truths in the universe for the hypotheses would be revealed. However, it is difficult and inefficient to do so, and is, in fact, impossible in most cases. There are even cases in which studies for the entire population turn out to be more inaccurate than studies conducted over a sample. Therefore, researchers select samples to represent the complete set of subjects of the study, and must analyse the data of the sample in order to extrapolate the characteristics to the whole population.
The general population refers to all the entities of interest in the study. By imposing clinical and demographic limitations, the study population, or target population, is selected. The study population is used to draw answers to the questions raised in the study. It is a collection of subjects representing basic units that are expected to be affected by the therapy or treatment studied in the research. In general, it is not easy to conduct sampling that represents the study population perfectly, due to the following realistic problems. The accessible population is the collection of subject units that can be sampled and studied. The accessible population is determined by applying environmental, time, and locational limitations on the study population. This requires specification of the research period and the medical institution in which the research is to be conducted. The study's degree of systematization, experience, resources, and underlying risks depends on the level (primary, secondary, and tertiary) of the hospital in which it is conducted and whether or not it is conducted in a local community. A number of sociological, economic, cultural, and climatic factors may also affect the study's external validity. The readers and reviewers are interested in whether or not the population of the research represents the general population, and whether the research results are suitable and applicable to the field of interest. Therefore, it is necessary for the researchers to provide information about the factors that may affect the external validity of the study (including the location, environment, and time). After defining the subject population using eligibility, inclusion and exclusion criteria, the research should proceed through sampling (Fig. 1). Understanding the overall process of the research is important to evaluate how the research results are relevant and to what kind of people they are applicable. Therefore, the process must be described thoroughly in the literature.
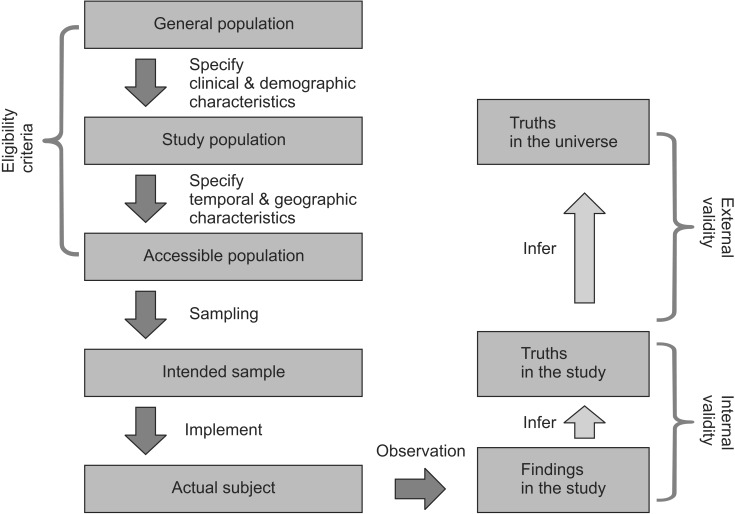
Illustration of study.
3. Is the sampling method specified?
Sampling methods are divided into probability and nonprobability sampling methods, depending on whether or not every unit in the population has the same chance of being selected in the sample. For the probability sampling method, the probability of sample selection is accurately known before the sampling. Therefore, it can be used to calculate the statistical reliability of the population estimator drawn from the sample data probabilistically. The nonprobability sampling method may be influenced by surveyor's biases, making the sample inadequate in representing the population. Despite this shortcoming, the method is used in cases in which the population cannot be defined accurately, in which the sampling error is not important, or in which a probability sampling method is not required. The nonprobability sampling method is often used as it is both economical and convenient. However, in those cases, there should be an assumption that the traits of the sample can easily be found to represent those of the population. Furthermore, the sampling must be carried out as similarly to the probability sampling method as possible. Randomization should be used to compare different groups. In medical research, two nonprobability sampling methods-convenient and consecutive-are generally employed without mentioning them in the papers. Articles without specification of sampling methods make it hard to evaluate the accuracy of the estimation. They are also limited, in that the criteria and results of the research are subject to change depending on the researchers and the environment. Moreover, they are prone to being influenced by researchers' subjectiveness. Therefore, this information must be transparently communicated to readers and reviewers.
4. Does the sample adequately represent the study population? (Is there external validity or consideration of external validity?)
Validity refers to the degree to which the conclusions or statistics (drawn from sample) reflect the true value (estimator drawn from population). There are two types of validity: internal and external. Internal validity reflects whether the outcomes result from the experimental treatment. It proves that they did not occur by accident, bias, or through confounding variables. It is reasonable to say that a study is internally valid if its RCTs have been carried out without any significant methodological problem, and if every considerable issue has been taken into account. Internal validity is a prerequisite of external validity-a study without internal validity cannot have external validity. External validity-also called generalizability or applicability-is concerned with whether or not the results can be applied to the general population. A study is considered to have external validity when it is conducted in an environment similar to the real one [12] (Fig. 1). Researchers may be keen to argue that their theories drawn from and proven by randomized controlled trials can be applied not only to participating patients, but, more generally, to all patients. In other words, they may hope that their research results can be generalized to the study population and, beyond, to the general population. In order for this to happen, a judgment needs to be made of the extent to which the results can be generalized in terms of time and space [13]. There should be information in the paper about the extent to which the participating patients represent the entire group of patients. It is also important to specify the factors that may prevent the results from being generalized to all patients. Although randomized clinical studies may have high internal validity, it should not be overlooked that they may have low external validity due to the high risk that the research participants might not represent the general population.
5. Are there grounds for statistical power or sample size calculation?
An accurate calculation of the statistical power or sample size is essential to conduct efficient and ethical research without wasting unnecessary cost and effort. It also gives researchers more chances of observing the effects of the intervention [14]. In many cases, researchers determine the sample size randomly or only in as much as the given resources allow, which is illogical even unethical. On the other hand, an unnecessarily large sample size wastes the limited time, cost, and resources. If the sample size is larger than needed, the research subjects may be exposed to unnecessary risks. Moreover, these cases may cause not only the desired evaluation variables but also other variables to look significant, which could hinder researchers from identifying the variables whose validity really needs to be evaluated. In contrast, a less-than-necessary sample size may result in a statistical power insufficient to discover significant differences. The research outcome may not have any practicality if the sample size is too small, even though its significance could have been proven and applied in the medical field if it had been observed with an adequate sample size. On the other hand, the effectiveness of an intervention may be overrepresented in cases in which a small sample is used [15], which can hurt the reliability of the study's conclusions [16]. Therefore, statistical power or sample size calculation must be taken into consideration before conducting randomized controlled trials, and must be specified in the paper.
The description of the sample size calculation should be detailed enough for readers and reviewers to be able to reproduce it. The information given should include the statistical power; the significance level; the clinically meaningful difference; and the ratio, mean, and standard deviation of the expected values from the control group and the treatment group. The statistical power must be at least 80 percent, and the clinically meaningful difference should be significant enough to be accepted clinically and should not be manipulated into a larger figure in order to reduce the sample size. Therefore, the sample size should be carefully planned according to balanced clinical and statistical considerations [17].
Generally, the sample size calculation and statistical power analysis should be based on the primary endpoint. One must remember that sample size calculations and power analyses based on primary endpoints may not be applied to secondary end-points. Furthermore, if there are multiple primary endpoints, the number of hypotheses may be increased to match the number of primary endpoints. Therefore, the sample size estimation should be carried out after correcting for the type I error with a multiple range test. Bonferroni's (P = mp) and Sidak's (P = 1 - (1 - p)m) methods are the most commonly-used correction methods [17].
6-1. Is the randomization explained?
6-2. Is the sequence generation method explained? (e.g., whether the application of block or who conducted the sequence generation)
7. Is the allocation concealment method explained?
Randomization refers to the assignment of subjects to each group through a stochastic process that is not influenced by the researcher's opinion [18]. Through randomization, researchers can make each group similar not only in terms of known prognostic factors such as the severity of the disease, age, and gender, but also of others less obvious or unknown. This provides them with a basis for arguing that any differences among the groups are due to differences in treatment. Randomization eliminates the biases that may have occurred if the researchers had done the assignment themselves, providing statistical grounds for arguing that the differences in research outcomes must result from differences in treatment, given that they were probabilistically highly unlikely. Furthermore, since most statistical methods are based on probability theories, randomization confers validity to the statistic test [17]. As mentioned before, most research uses consecutive or convenient sampling methods, which are nonprobability sampling methods. Therefore, randomization is highly necessary, and any randomization method employed in the research should be explained in the paper.
Randomization largely consists of three processes: random sequence generation, allocation concealment (which is particularly important when carrying out research), and commencement.
Random sequence generation is the process of generating sequences by chance and by making the group assignment unpredictable. Research papers should provide enough information about the process for readers and reviewers to be able to evaluate whether the sequence generation process was biased or not. Rotating assignment, or assignment based on the patient's chart number, birthday, date of visit, or examination result, cannot be considered as random sequence generation. However, many studies employ these methods and use the word "random." In those cases, the assignment method should be clarified and should not be called "random." Some reports claim that research without randomization is often biased [4,19]. Certain elements must be specified when randomization is used: the kind (simple, block, stratification, etc.) of randomization; the methods used to generate and operate randomization list (table of random numbers, computerized generation of random numbers, coin toss, etc.); the allocation ratio; and whether the randomization was stratified (Table 2).
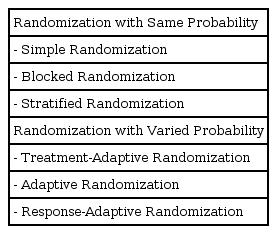
Type of Randomization
Randomization should be used in conjunction with allocation concealment [20]. Allocation concealment refers to a condition according to which no one knows how the patients will be allocated at the time of recruitment. If a researcher is aware of groups the patients will be allocated before the recruitment, he or she may be consciously or unconsciously influenced when deciding whether certain patients should be allowed to participate in the study or not. Allocation concealment prevents these issues. Whether a patient should be recruited as a study subject or not must be determined before the allocation [21].
Allocation concealment is not to be confused with blindness, which will be explained below. Allocation concealment makes sure that no one knows what each group will look like before the allocation, and blindness is concerned with confidentiality after the allocation. The former prevents selection biases, and the latter prevents performance and measurement biases (Fig. 2). Allocation concealment can be successfully applied to any type of randomized controlled trials, while blindness is not always possible [20]. For example, in studies comparing a short arm splint and a long arm splint, it is impossible to use blindness, as the patients and researchers cannot be prevented from knowing who was assigned to which group after the allocation, since every participant is bound to receive information following the intervention. However, it is possible to keep information about the group allocation undisclosed before recruiting the participating patients. If a researcher is aware of who will be allocated to which group before recruiting the participants, there is a higher risk that he or she, whether consciously or unconsciously, will assign patients who are expected to show positive findings to the treatment group so that the study may turn out to be more significant.
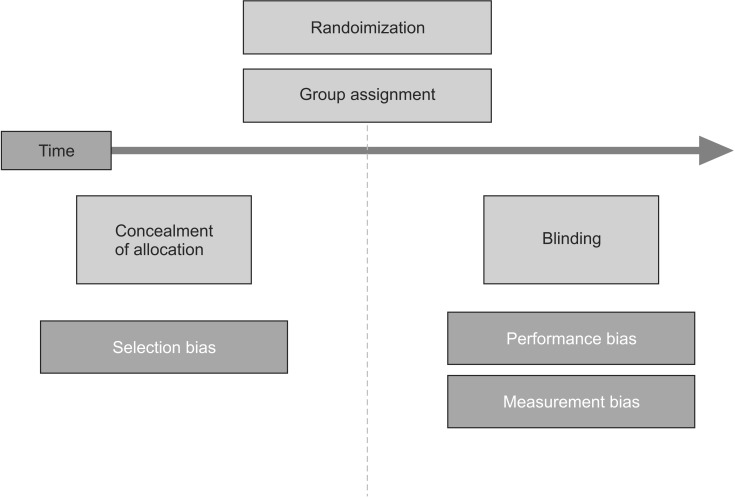
Comparisons of allocation concealment and randomization.
The person responsible for allocating the patients must not be aware of the allocation sequence generation and the allocation concealment. Therefore, the ideal way is to exclude the person in charge of the processes from the process of allocation of the patients to the groups. It is therefore important to provide information in the paper about who generated the allocation sequence, who recruited the participating patients, and who allocated them to each group. Allocation concealment is one of the most powerful experiential forms of evidence affecting the research results. According to a report, studies with inadequate or misapplied allocation concealment tend to produce results that exaggerate the effects of the intervention [4].
8-1. Does the research employ blindness and give explanations for it?
8-2. Is the blindness acceptable?
In experimental research, blindness is used to reduce the biases that may occur when conducting experiments or evaluating results. Blindness is particulary important for the results of subjective variables [22]. It may also affect the intervention compliance and the dropout rate of the study.
A single-blind procedure refers to making patients unaware of which treatment they are receiving. In other words, the patients are not informed of whether they are in the control group or the treatment group. The single-blind procedure is used on account of the fact that the evaluation of the treatment's effectiveness may be influenced by placebo effects if the participants are aware of which intervention they have received and are psychologically affected. A double-blind procedure prevents not only the participants but also the researchers who observe and analyze the outcomes from knowing which patient belongs to which group. In a clinical trial, blindness should be applied to the researchers, doctors who are not researchers, examiners, data collectors, and other hospital staffs such as nurses, pharmacists and medical engineers, who are all in direct contact with the participating patients [23,24].
The odds ratio of studies conducted without blindness was found to be exaggerated by 17 percent [4].
However, unlike allocation concealment, blindness cannot be applied on every occasion. For example, in research comparing pain after Laparoscopic and Open Cholecystectomy, it is impossible to apply blindness to the surgeons. However, it may be possible to apply it to the persons in charge of measuring the degree of pain, as well as others involved in research. Therefore, blindness should be used to the extent that will seem reasonable to the readers and reviewers.
Regardless of the degree of blindness, the blindness subjects (i.e., the research subjects, researchers, doctors, data collectors, examiners, etc.) must be disclosed accurately. It is particularly important to disclose whether blindness was used on the research subjects, the interviewers, and the evaluators. Where possible, the traits (shape, taste, odor, and administration method) and similarities in the treatments should be explained as well [25]. This is because, despite the blindness, patients, examiners, or data collectors may realize which intervention is being used in the course of the administration, which may influence the research results. Some researchers tested whether blindness had worked properly by surveying the people under the influence of blindness about the patient allocation after the research [26].
The CONSORT Check List and other references have already emphasized the importance of describing the blindness used in research6), [27,28,29]. It is hard for readers and authors to evaluate the validity of studies unless authors accurately explain the blindness used in those studies.
9-1. Is a list of variables accurately provided? Are the variables of interest clearly defined?
9-2. Are there explanations of the variables that are rarely used?
Every randomized controlled trial features variables or values that can be compared. Most research includes a multiple number of variables, and researchers may be more interested in certain variables than others. The primary outcome variable is the one the researcher is most interested in, and is considered as the most important variable in the study. The primary outcome variable is also used to calculate the sample size. Some studies may contain more than one single primary outcome variable, although this is not recommended as multiplicity may occur. If more than one primary outcome variable is present, researchers must fully consider the issues that may arise from multiplicity. Secondary outcome variables refer to outcome variables other than the primary outcome variable. There may be more than one secondary outcome variable. Since studies with multiple outcome variables have a higher risk of reporting the results selectively, researchers must designate and report the outcome variables in a research protocol and registry before initiating the clinical trials, and must specify them in the reports and papers.
According to a study that compared the research protocols and published studies approved by independent review boards, 62 percent of them showed different primary variables in the protocols and published papers, as researchers either created new primary variables, or changed or omitted the original ones [30]. A systematic review of the published research leads to a similar conclusion [8].
Every variable must be defined accurately, and, as in the case of information about the intervention, the information should be provided to everyone who is susceptible of making use of the outcome variables. Reviewers and readers should be provided with an extensive explanation of any variable that is not commonly used. Related studies should be given for variables and scales that have been used and have been proven to be effective in prior research. Using a proven scale is highly recommended, as a scale without proven effectiveness may cause an exaggerated result [5].
If the measurement of the results is conducted at multiple time points, researchers must designate the most meaningful time point, or clarify whether they want to observe the differences in specific time point or the overall differences.
Commencement of Trial
1-1. Does the paper describe the interventions applied to the compared groups and details them, including the time and method, to the extent that other researchers can reproduce them?
1-2. Does it clearly explain the method for measuring the results?
Researchers must comprehensively explain the interventions applied to each group, including those applied to the control groups. The explanations must be detailed enough for other clinicians who want to use the studied interventions to reenact them [31]. For interventions using a drug, the name of the drug, the amount used, the administration method, the administration time and period, and the conditions for cessation must be provided. As the method used to measure the results can cause biases, the researchers should also elaborate on this issue.
2. Are the differences between the research proposal and the actual result mentioned?
Any change that occurred after the initiation of the research must be mentioned in the report. There are cases in which a study cannot be, or is not, conducted as planned. These may include cases in which another study or a systematic literature review reports that the primary variable is not adequate, or in which it is concluded that an intervention may harm patients. It may be unethical to continue with the research if an outcome variable turns out to be directly threatening to the safety of the patients. Another case may be when there are not enough or too many patients recruited in the study. When the plan for the research changes, as in the cases mentioned above, the changes must be reported to the registry and specified in the paper. Moreover, any unintentional eligibility criterion, intervention, test, data collection, result, or analysis method used must be made transparent in the report.
3. Does the paper report the side effect, reaction, and harm variables?
There are variables related to effectiveness and harm. Harm variables carry much weight and must be reported regardless of whether they are primary or secondary. Readers and reviewers want to be able to make conclusions that are reasonable and balanced. Therefore, they need to know not only about the benefits but also about the harm that may be caused by the interventions. However, not every harm outcome is usually reported. In fact, a number of studies have revealed that a lot of abnormal reactions and information related to safety go unreported [7,32,33].
4. Is information about the patients who dropped out from the treatment group and the control group included? Is the follow-up rate high?
Some studies proceed as planned from the beginning, without any subject dropping out or being excluded from the study. Although these cases involve no surprises, this should nevertheless be reported. For some research featuring complex outlines, readers may find it hard to clarify whether some patients did not receive the planned interventions, dropped out in the course of the research, or were excluded from the analysis after receiving the intervention [34].
This information is critical, given that if a patient dropped out after the intervention, he or she may be inadequate to represent the population and might have experienced a worsening of the symptoms or unexpected side effects [35]. It is not rare to see patients drop out of a study. However, it should be clearly stated whether the patient dropped out of the study voluntarily, whether the researchers decided to exclude the patient because he or she was found not to meet the subject criteria, or whether the patient failed to follow the research plan. This is important as this information provides grounds for different interpretations of the study, especially in relation to biases [35,36,37].
For example, in a study that compared the effectiveness of carotid endarterecomy and medical treatment in patients that could receive follow-up, the patients who had received carotid endarterecomy showed a significant decrease in the number of ischemic strokes and deaths [38]. However, when the result was reanalyzed using an intention-to-treat method, it lost its significance or showed less effectiveness [35].
It is also important to record the number of eligible patients [39], as this can help to assess whether the sample used in the research represented them adequately. However, many randomly-controlled trials do not provide sufficient information. According to a study, 20 percent of randomly-controlled trials did not report on the randomly-allocated people and those excluded from the research [34].
The CONSORT statement recommends using a flow diagram7), to show the patients who were not included in the conclusion [40].
Conclusion
Studies involving randomly controlled trials (RCTs) provide credible information about the effectiveness of an intervention. This explains why many clinical trials employ RCTs. However, many researchers do not consider or leave out important issues involved in RCTs from their reports.
When choosing the topic of a study and when planning and initiating it, it is important to make sure that the research has internal and external validity. To avoid biases and enhance the study's internal validity, researchers should adopt an adequate methodology. The sample size is also important for obtaining an appropriate statistical power. The research subjects should be able to represent the general population.
A study may not be published, or may be considered to be biased by readers and reviewers after publication, if it has left out important information in the report despite being otherwise properly conducted. Therefore, it is recommended that researchers refer to guidelines when reporting on their studies.
While this guideline may not be perfect, we hope that it will help researchers design better studies and reports, and will contribute to the advancement of the Korean Journal of Anesthesiology.