Inhaled nitric oxide for the brain dead donor with neurogenic pulmonary edema during anesthesia for organ donation: a case report
Article information
Abstract
Neurogenic pulmonary edema (NPE) in brain dead organ donors occurring after an acute central nervous system insult threatens organ preservation of potential organ donors and the outcome of organ donation. Hence the active and immediate management of NPE is critical. In this case, a 50-year-old male was admitted to the intensive care unit (ICU) for organ donation. He was hypoxic due to NPE induced by spontaneous intracerebral hemorrhage and intraventricular hemorrhage. Protective ventilatory management, intermittent recruitment maneuvers, and supportive treatment were maintained in the ICU and the operating room (OR). Despite this management, the hypoxemia worsened after the OR admission. So inhaled nitric oxide (NO) therapy was performed during the operation, and the hypoxic phenomena showed remarkable improvement. The organ retrieval was successfully completed. Therefore, NO inhalation can be helpful in the improvement of hypoxemia caused by NPE in brain dead organ donors during anesthesia for the organ donation.
Brain death brings upon significant physiological instability in the cardiovascular, respiratory, and endocrine systems. Poor management of a brain dead patient scheduled for organ donation can cause deterioration of the organ functions prior to retrieval, which may make the organ transplantation not viable [1]. Therefore, it is important for the patient to have active treatment from the point of declaration of brain death to the point of organ retrieval that optimizes the donor's organ perfusion, stabilizes endocrinal homeostasis, and protects weak organs, since the quality of the management of the donor patient greatly affects the organ functions and the prognosis of the recipient [1,2]. Among common physiological abnormalities in brain dead organ donors, the incidence of pulmonary edema comprises 13-18% [1]. NPE is characterized by acute pulmonary edema occurring after a central nervous system (CNS) insult without underlying cardiovascular or respiratory pathology [3,4]. It causes severe hypoxia and can threaten organ preservation of the potential organ donor [5]. Therefore when NPE occurs in a brain dead organ donor, expeditious treatment is necessary for the improvement of hypoxia. The authors present a case of a brain dead organ donor with NPE whose hypoxemia became exacerbated despite continuous protective ventilatory management, intermittent alveolar recruitment maneuvers, and supportive treatment of the patient during anesthesia for the organ donation. Thus NO inhalation was performed, which remarkably improved oxygenation and successfully allowed the completion of the organ retrieval surgery.
Case Report
A 50-year-old male patient without spontaneous respiration and with pupils dilated and fixed at 7 mm was admitted to the ICU. The patient had no past history, but 8 days prior to being admitted, he had received an emergency decompressive craniectomy and hematoma removal at a different hospital for spontaneous intracerebral and intraventricular hemorrhage. But there was no neurological recovery, so a follow-up brain CT was performed, which showed rebleeding. A second surgery was performed. After treatment in the ICU, he was transferred to our hospital for organ donation upon brain death.
Upon being admitted into the ICU, the patient's vital signs were blood pressure (BP) 75/50 mmHg, heart rate (HR) 72 bpm, and pulse oxygen saturation (SpO2) 75%. Fluid administration and continuous intravenous infusion of dopamine 15 µg/kg/min were started. For mechanical ventilation support, pressure control ventilation was started with fraction of inspired oxygen (FiO2) 1.0, peak inspiratory pressure (PIP) 26 cmH2O, positive end expiratory pressure (PEEP) 10 cmH2O, and respiratory rate (RR) 20 breaths/min. General edema of the patient and a large amount of pink frothy sputum in the tracheal tube were observed. When the lungs were auscultated, rales were broadly audible bilaterally. A chest X-ray showed bilateral infiltration (Fig. 1). On echocardiography, concentric left ventricular hypertrophy with preserved contractility was observed, but there was no evidence of pulmonary arterial hypertension. The first arterial blood gas analysis (ABGA) findings upon being admitted into the ICU (FiO2 1.0) were pH 7.310, PaCO2 40.5 mmHg, PaO2 84.9 mmHg, HCO3- 19.9 mEq/L, base excess (BE) -5.9 mEq/L, and SaO2 94.3%. Besides the CNS insult, there was no other existing cause for the pulmonary edema such as heart failure, pneumonia, or pulmonary embolism. So it is believed that the NPE from the intracerebral and intraventricular hemorrhage caused the hypoxemia.
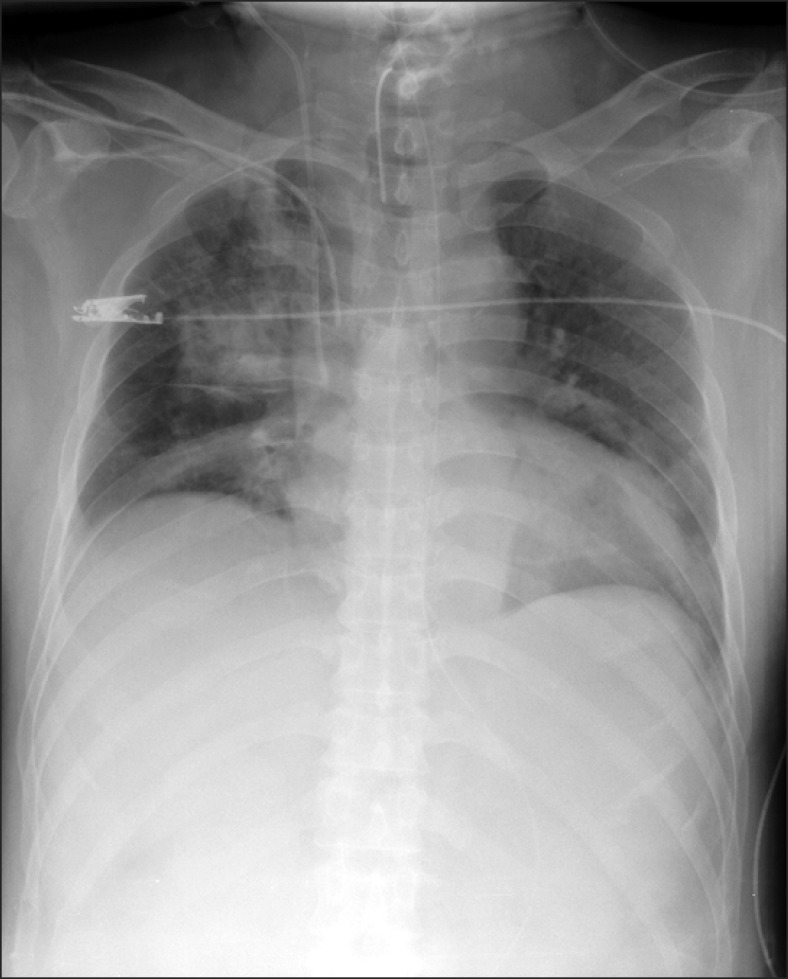
Chest radiograph obtained on admission to the intensive care unit. There is bilateral diffuse infiltration, which is suggestive of pulmonary edema.
In the ICU, the ABGA findings immediately after the first apnea test decreased to SaO2 85.7% and PaO2 66.7 mmHg. For 2 hours around the time of the first apnea test, SpO2 intermittently decreased to 91-93%. Each time it dropped, alveolar recruitment maneuvers were performed using a continuous positive airway pressure (CPAP) of 35-40 cmH2O for 30 s with BP monitoring, with the RR maintained at 15-20 breaths/min, and PEEP raised to 15 cmH2O. So the patient's SaO2 was maintained at 96.3% or above and the PaO2 at 93.4 mmHg or above in the ICU. A large amount of frothy sputum was continuously removed intermittently by endotracheal suction. By the continuous intravenous infusion of dopamine 15 µg/kg/min, the patient was maintained at a systolic pressure of 90-120 mmHg, diastolic pressure of 50-80 mmHg, HR of 80-95 bpm, and central venous pressure of 11-13 mmHg. The patient had accompanying diabetes insipidus, metabolic acidosis, hyperglycemia, and hypernatremia, and so he was treated with fluid supplementation, electrolyte correction, sodium bicarbonate administration, and a continuous intravenous infusion of insulin for the management of the blood glucose. A warm air blanket was used to maintain the body temperature at 36℃ or above. The total amount of fluids administered in the ICU was 6,280 ml of crystalloid solution and 80 ml of colloid solution (20% albumin). The total urine output was 3,840 ml, and the fluid amount drained from the nasogastric tube was 1,300 ml. The patient showed a loss of brainstem reflexes, and two apnea tests taken at 6-hour intervals, a transcranial Doppler, and an electroencephalography showed findings congruent with brain death, so brain death was declared. After 10 h and 30 min in the ICU, the patient was transferred to the OR for organ retrieval. During the transfer, oxygen 12 L/min was supplied to maintain the FiO2 at 1.0, and manual ventilation was maintained using a resuscitation bag with a reservoir bag.
Before the patient was transferred, the last vital signs measured were BP 120/70 mmHg, HR 75 bpm, and SpO2 99%. Upon arrival at the OR, the vital signs were BP 87/55 mmHg, HR 100 bpm, and SpO2 80%. The anesthesia machine was connected, an inhalational agent was not used, and mechanical ventilation was started with the same settings used in the ICU. The SpO2 slowly rose, but despite intermittently performing alveolar recruitment maneuvers and raising the PEEP to 15 cmH2O and the PIP to 30 cmH2O, the SpO2 did not rise to more than 91%. At this point, the ABGA findings (FiO2 1.0) were pH 7.412, PaCO2 33.0 mmHg, PaO2 62.3 mmHg, HCO3- 20.5 mEq/L, BE -3.6 mEq/L, and SaO2 90.8%. To raise the BP to the target systolic pressure of 100 mmHg or above and the mean arterial pressure to 70 mmHg or above, dopamine 15 µg/kg/min was maintained and epinephrine 0.05 µg/kg/min was intravenously administered continuously in addition. However, even when the BP rose to the target level and the surgery was proceeded with, the SpO2 did not rise any further. Therefore, NO gas inhalation as an adjuvant therapy was decided upon. After 20 min from the beginning of the operation, a NO infusion was started at 20 ppm using the inspiratory limb of the ventilator and the level was monitored so that the NO2 concentration did not go over 5 ppm. Within a few minutes after the NO infusion, SpO2 rose from 91% to 99%. ABGA performed 20 min after the NO infusion showed pH 7.270, PaCO2 46.3 mmHg, PaO2 328.7 mmHg, HCO3- 20.8 mEq/L, BE -6.0 mEq/L, and SaO2 99.5%. The hypoxemia had significantly improved. Afterwards, NO administration was continued at 20 ppm until the end of the anesthesia. Follow-up ABGAs showed the maintenance of SaO2 at 98.4% and above and PaO2 at 132.4 mmHg and above (Table 1). During surgery, supportive treatment for the blood glucose, electrolytes, and acidbase balance was continued in the same manner as in the ICU. Intermittent endotracheal suctioning was continued to remove a large amount of frothy sputum, and muscle relaxation was maintained by the intravenous administration of vecuronium. During the 3 h and 5 min of anesthesia time, the total amount of fluids and blood administered were crystalloid solution 2,380 ml, colloid solution 500 ml, and red blood cells 4 units. The estimated blood loss amount was 1,300 ml, urine output 1400 ml, and the drained fluid from the nasogastric tube 340 ml. Within these figures, the total amounts of fluids and blood administered until the beginning of NO inhalation were crystalloid solution 570 ml and red blood cells 0.5 units, and the estimated blood loss amount was 150 ml, urine output 320 ml, and the drained fluid from the nasogastric tube 80 ml. As soon as cross-clamping of the aorta was performed 2 h 40 min from the beginning of the operation, the patient's BP and SpO2 suddenly decreased. An electrocardiogram showed arrhythmia, bradycardia, and then cardiac arrest, and the brain dead patient was declared dead. The surgery was completed after two kidneys were uneventfully retrieved for the transplantation to two organ recipients.

Arterial Blood Gas Analysis Before and After NO Inhalation
Discussion
NPE is a clinical syndrome where pulmonary edema occurs shortly after a serious CNS insult. Usually it occurs without a cardiovascular or respiratory etiology that is the usual explainable cause of pulmonary edema. However, it does not exclude a coexisting effect of neurological damage on myocardial function, and a pre-existing myocardial dysfunction can cause confusion about the occurrence of NPE [3]. There are many reported causes of NPE such as subarachnoid hemorrhage, traumatic brain injury, intracranial hemorrhage, epilepsy, meningitis, subdural hemorrhage, cervical medulla injury, cerebral thrombosis, cerebral gas embolism, medication overdose, multiple sclerosis, and arteriovenous malformation [3,4,6]. Symptoms of NPE occur most commonly within minutes to hours following the neurologic injury. Although there is a 12-24 h delay in manifestations, the rapid development of respiratory failure is an impressive feature. The clinical presentation appears as signs of respiratory failure. Dyspnea, tachypnea, tachycardia, cyanosis, and pink frothy sputum are often observed. Upon pulmonary auscultation, bilateral crackles and rales can be heard. ABGAs show a low PaO2 and a decreased PaO2/FiO2 ratio below 200. On chest X-rays, bilateral diffuse alveolar infiltrations are usually observed. As a differential diagnosis, the possibility of aspiration pneumonia that is common among CNS injured patients must be excluded. There must not be the presence of other common causes of acute respiratory failure such as a massive blood transfusion and sepsis. The possibilities of post-airway obstruction edema, ventilator-associated pneumonia, and ventilation-induced lung injury must also be ruled out [3,4,6,7].
The patient in the present case had no past history, and the echocardiography did not show findings that would have caused cardiogenic pulmonary edema. 8 days before the patient was admitted to the hospital, severe spontaneous intracerebral and intraventricular hemorrhage had occurred. In the previous hospital the patient was at, there was no aspiration during endotracheal intubation. There was no blocking in the endotracheal tube. Tidal volume and airway pressure were controlled with a protective ventilation strategy similar to the management of an acute lung injury. All of the above factors as well as the patient's clinical signs, findings on the chest X-ray and ABGA led to the assumption that NPE had occurred.
The pathophysiology of NPE has not been clearly established. Two different mechanisms seem to coexist, triggered by a sudden elevation in intracranial pressure and a global decrease in cerebral blood flow or localized ischemia in the NPE trigger zones. NPE trigger zones are mainly found in the hypothalamus and medulla oblongata [8]. Firstly, the hemodynamic mechanism, which is usually called the blast injury theory, starts with a sudden increase in the intracranial pressure, which causes a sudden increase in catecholamines and causes both systemic and pulmonary vasoconstriction. This increases the pulmonary hydrostatic pressure, which subsequently increases the permeability of pulmonary capillaries. Secondly, the inflammatory mechanism states that cytokines which are released due to the brain injury increase the permeability of pulmonary capillaries. These mechanisms make the fluid shift into the pulmonary alveoli and the interstitial space, which leads to pulmonary edema [3]. The respiratory pathophysiology causing pulmonary edema in the brain dead patient follows a similar mechanism [1,2].
The protective ventilatory strategy for brain dead organ donors is similar to the ventilatory management of patients with an acute lung injury or acute respiratory distress syndrome, and this management also applies to brain dead organ donors with NPE [3]. Lung units are retained by applying a PEEP of 5 cmH2O while limiting the tidal volume to 4-8 ml/kg of the ideal body weight and the plateau pressure to 30 cmH2O with a focus on recruitment. Re-recruitment is important especially after tracheal suction or after the apnea test. PEEP can be increased up to 15 cmH2O without impeding cerebral perfusion pressure [1,2,3]. In the treatment of NPE patients, the generally known supportive treatment includes endotracheal intubation, sedation, supplemental oxygen, moderate PEEP administration, and vasoactive drugs, diuretics, steroids, and fluid supplements [3,4,6,7]. An extremely high increase in the level of catecholamines in NPE patients due to lung injury can cause hemodynamic instability and respiratory failure. There is a report on the intravenous administration of the α-adrenergic blocker phentolamine improving the clinical status, but further studies on the improvement are needed [9]. Hemodynamic instability leads to peripheral organ hypoperfusion, which worsens metabolic acidosis and the progress of inflammation and causes an increase in multiorgan failure. Therefore the use of vasoactive drugs may improve local perfusion and slow down the inflammatory process [3].
According to Salim et al. [10], the implementation of aggressive management of organ donors raised the number of potential organ donors and actual donors respectively by 19% and 87% when compared with the period before aggressive donor management. It also reduced the loss of donors due to hemodynamic instability by 87%. Also, it increased the number of retrieved organs by 71%. In addition, a study on lung transplantation stated that a low donor PaO2 is correlated with a low recipient PaO2/FiO2 ratio (P = 0.007), and this raises the incidence of prolonged postoperative mechanical ventilation, and predicts early graft dysfunction and hypoxia [11]. Although the organs donated in the present case were the kidneys, not the lungs, this fact points to the importance of active management of the brain dead patient, including maintenance of optimal oxygenation when an organ donation is scheduled. Therefore, according to the cardiovascular goals for the active management of potential organ donors suggested by Shemie et al. [12], active ventilatory management was performed to maintain ABGA pH at or above 7.35, PaCO2 between 47 and 60 mmHg, PaO2 at or above 107 mmHg, and SaO2 at or above 95%. Nevertheless continuous hypoxia had to be corrected by initiating NO inhalational therapy. Also vasopressors were administered by a continuous intravenous infusion to keep the systolic pressure at 100 mmHg or above, mean arterial pressure 70 mmHg or above, and HR 60-120 bpm. Supportive treatment was given, with a target of maintaining adequate levels of urine output, electrolytes, blood glucose, and normal levels of other physiological parameters.
The purpose of trying NO inhalation in the present case was as follows: firstly, the SpO2 in the ICU dropped to 91-93% intermittently. Each time it dropped, the SaO2 was raised relatively easily by RR control, a PEEP increase, and an alveolar recruitment maneuver using CPAP. At other times, the patient's SpO2, SaO2, and PaO2 in the follow-up ABGA were maintained near the target levels. Immediately after the OR admission for organ donation surgery, the SpO2 dropped to 80%. This drop may have occurred despite the performance of manual ventilation during the transfer with a resuscitation bag using an attached reservoir bag to maintain the FiO2 at 1.0 because there was not enough PEEP. But afterwards, mechanical ventilation with the same settings that were used in the ICU and the treatment still did not raise the SpO2 to more than 91%. There was a limit to the improvement of hypoxemia. The level of PEEP and plateau pressure applied to the patient had been increased to the maximum level recommended for protective ventilation strategy. It was feared that a further pressure increase would raise the intracranial pressure and central venous pressure, decrease venous perfusion and cardiac output, and cause ventilator-induced lung injury. So NO gas inhalation was decided upon. Secondly, after connecting a ventilator to the patient, the SpO2 rose to 91%, and the additional intravenous infusion of epinephrine made the BP reach the target level. It was then feared that the donor's condition would worsen with the passing of time, so the surgery was proceeded with without delay. Extracorporeal membrane oxygenation could have been considered, but rather than delaying the progress of the surgery which had already started, for the purpose of cannulation, NO inhalation was preferred and decided upon because it is noninvasive, and easily and rapidly performed. Fortunately, NO inhalation therapy proved to be successful.
NO inhalation causes the selective dilatation of vessels associated with the ventilated lung units that are reached by the inhaled gas diffusing across the alveolar-capillary membranes. So it results in preferential pulmonary vasodilatation without affecting the systemic BP, reduces pulmonary vascular resistance, augments hypoxic pulmonary vasoconstriction, and improves oxygenation and ventilation-perfusion matching. Therefore, NO inhalation is used in the treatment of acute lung injury and acute respiratory distress syndrome patients. But the licensed indication of NO inhalation is restricted to persistent pulmonary hypertension in neonates. Also, there is not enough evidence concerning any benefits in the outcomes [13,14]. Yet patients with acute hypoxia and life-threatening pulmonary hypertension can have NO inhalation therapy as a short-term adjuvant therapy [13]. NO inhalation therapy usually starts at 5-20 ppm, and if there is no reaction within a few minutes, it is raised up to 80 ppm. But during chronic NO inhalation therapy, it is recommended that less than 40 ppm be administered in clinical practice. At such clinical doses, methemoglobinemia should not occur, but regular checks of methemoglobin levels are still recommended. NO2 production during NO inhalation is from NO binding with O2 and occurs when there are high concentrations of NO and O2 [14,15]. NO2 at 5 ppm or above causes pulmonary toxicity, so it is important to continuously monitor the levels of NO and NO2, and to minimize FiO2 using the minimum effective dose when possible. However, there are not many reported cases of pulmonary toxicity occurring from short-term exposure [15]. A sudden weaning in NO inhalation treatment has the risk of causing rebound pulmonary hypertension and severe hypoxia, so it is important to allow for slow weaning [13,15].
The patient in the present case inhaled 20 ppm NO and showed significant improvement in oxygenation. PaO2 and PaO2/FiO2 ratio were best at the first ABGA done 20 min after NO inhalation, but they became reduced with the passing of time. The pressure-controlled ventilation set at the maximum threshold of the protective ventilation strategy was maintained during the surgery, but the intraoperative respiratory acidosis and metabolic acidosis based on the BE value eventually worsened. It is believed that this was due the unstoppable exacerbation of the brain dead patient's pathophysiology despite active ventilatory management. A high concentration of oxygen (FiO2 1.0) encourages the production of NO2, but NO was inhaled for a short term with a clinical dose, with NO2 concentration monitoring. So it is believed that NO-induced pulmonary toxicity was not the cause of the gradually decreased improvement of the hypoxia. Unfortunately a chest X-ray was not taken after NO administration, which could have allowed for comparisons with the state before the NO was administered for prompt organ retrieval.
The benefits on outcomes of NO inhalation therapy are not clear. And it is not a standard treatment procedure. But there are many reported cases of patients with acute respiratory distress syndrome and pulmonary hypertension in the ICU and OR who benefited from NO inhalation. Also the clinical efficacy of NO is well-known. In the aforementioned cases, NO was used more in the ICU than in the OR. There are hardly any cases where NO inhalation therapy was performed on a brain dead patient scheduled for organ donation. There is just one case where 18 hours of NO inhalation improved NPE-caused hypoxia in the management of a brain dead patient in the ICU, and after stopping NO inhalation, anesthesia for organ retrieval was started [5]. Furthermore, there has been no reported case of successful surgery using NO inhalation therapy during anesthesia for organ retrieval in the OR, so the authors present this case. The present case shows that short-term NO inhalation therapy during anesthesia for organ retrieval in a brain dead organ donor with NPE improves hypoxia which was not improvable by only active treatment. Many organ donations are from brain dead patients with an acute CNS insult. So, in the management of brain dead patients, caution must be given to the possibility of the occurrence of NPE and great attention must be given to active ventilatory management and supportive treatment.