Polyphenol (-)-epigallocatechin gallate-induced cardioprotection may attenuate ischemia-reperfusion injury through adenosine receptor activation: a preliminary study
Article information
Abstract
Background
The activation of guanine nucleotide binding protein-coupled receptors, such as adenosine receptor (ADR) and opioid receptor (OPR), protects the heart against ischemia and reperfusion injury. We hypothesized that ADR or OPR might be involved in polyphenol (-)-epigallocatechin gallate (EGCG)-induced cardioprotection.
Methods
Langendorff perfused rat hearts were subjected to 30 min of regional ischemia and 2 h of reperfusion. Hearts were treated with 10 µM of EGCG, with or without the ADR or OPR antagonist at early reperfusion. Infarct size measured with 2,3,5-triphenyltetrazolium chloride staining was chosen as end-point.
Results
EGCG significantly reduced infarct volume as a percentage of ischemic volume (33.5 ± 4.1%) compared to control hearts (14.4 ± 1.1%, P < 0.001). A nonspecific ADR antagonist 8-(p-sulfophenyl) theophylline hydrate (27.1 ± 1.9%, P < 0.05 vs. EGCG) but not a nonspecific OPR antagonist naloxone (14.3 ± 1.3%, P > 0.05 vs. EGCG) blocked the anti-infarct effect by EGCG. The infarct reducing effect of EGCG was significantly reversed by 200 nM of the A1 ADR antagonist DPCPX (25.9 ± 1.1%, P < 0.05) and 15 nM of the A2B ADR antagonist MRS1706 (29.3 ± 1.7%, P < 0.01) but not by 10 µM of the A2A ADR antagonist ZM241385 (23.9 ± 1.9%. P > 0.05 vs. EGCG) and 100 nM of the A3 ADR antagonist MRS1334 (24.1 ± 1.8%, P > 0.05).
Conclusions
The infarct reducing effect of EGCG appears to involve activation of ADR, especially A1 and A2B ADR, but not OPR.
Introduction
Polyphenol (-)-epigallocatechin gallate (EGCG), a major catechin of green tea, by targeting ischemia [1] and reperfusion [2] provides cardioprotection against ischemia-reperfusion injury. We recently reported that the cardioprotective effect by EGCG was mediated via the ATP-sensitive potassium (KATP) channels [2].
On the other hand, homeostatic regulation and stress responses are mainly regulated by the extracellular signals transduced by guanine nucleotide binding protein (G-protein)-coupled receptor (GPCR) in the heart [3]. Adenosine receptor (ADR) and opioid receptor (OPR) belong to the GPCR family and activation of these upstream receptors might protect the heart by triggering second messengers [4,5].
We hypothesized that ADR or OPR might be activated by EGCG-induced cardioprotection. We therefore investigated the infarct reducing effect with ADR or OPR antagonists in EGCG-induced cardioprotection in isolated rat hearts.
Materials and Methods
The experimental procedures and protocols used in this study were reviewed and approved by our Institutional Animal Care and Use Committee.
Drugs and chemicals
EGCG, 8-(p-sulfophenyl)theophylline hydrate (8-SPT), and 2,3,5-triphenyltetrazolium chloride (TTC) were obtained from Sigma-Aldrich Chemical, St. Louis, MO, USA. Naloxone was purchased from Reyon Pharmaceutical Co., Seoul, Republic of Korea. Fluorescent polymer microspheres were purchased from Duke Scientific, Palo Alto, CA, USA. 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX), 4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo [2,3-a][1,3,5]triazin-5-ylamino] ethyl)phenol (ZM241385), N-(4-acetylphenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy] acetamide (MRS1706), and 1,4-dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3,5-pyridinedicarboxylic acid 3-ethyl-5-[(3-nitrophenyl)-methyl]ester (MRS1334) were purchased from Tocris Bioscience, Ellisville, MO, USA. Other chemicals were obtained from Sigma-Aldrich Chemical.
EGCG, 8-SPT and naloxone were dissolved in distilled water. DPCPX, ZM241385, MRS1706 and MRS1334 were dissolved in dimethyl sulfoxide. Stock chemicals were stored at -20℃ and were diluted with Krebs-Henseleit (KH) solution to the required final concentrations on the day of each experiment.
Langendorff isolated heart perfusion preparation
Male Sprague-Dawley rats, weighing 280-330 gm obtained from KOATECH Co., Cheongwon-gun, Republic of Korea, were used. They received 50 mg/kg of pentobarbital sodium and 300 IU of heparin intraperitoneally. Hearts were isolated and perfused with modified KH solution containing (in mM) 118.5 NaCl, 4.7 KCl, 1.2 MgSO4, 1.8 CaCl2, 24.8 NaHCO3, 1.2 KH2PO4, and 10 glucose, as described previously [6]. Regional ischemia was induced by pulling the snare which was made at the level of the proximal length of the left coronary artery (LCA) and its major branches and confirmed by regional cyanosis and a substantial decrease in left ventricular developed pressure (LVDP). Reperfusion was started by releasing the snare.
Experimental protocol
All hearts were subjected to 30 min of regional ischemia and 120 min of reperfusion. Infusion of EGCG and antagonists was started 10 min before the onset of reperfusion and continued for 40 min (Fig. 1). To assess the involvement of ADR or OPR in EGCG-induced cardioprotection, ADR and OPR antagonists were perfused via 2nd port 10 min before EGCG perfusion. The concentrations of all chemicals were based on our and other previous studies on isolated working rat hearts that had no effect on infarct size in hearts subjected to ischemia and reperfusion [5,7-11].
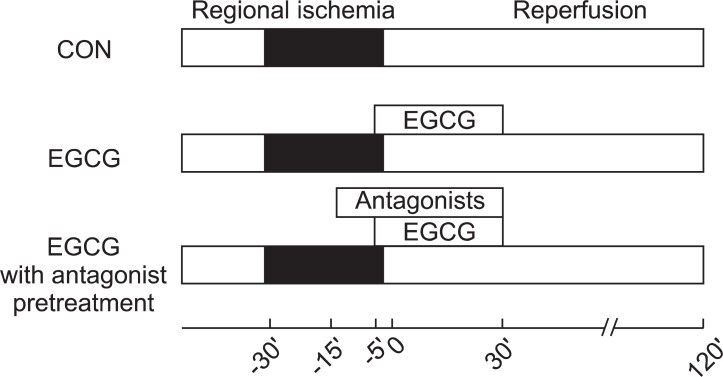
Experimental protocols. Hearts were subjected to 30 min of regional ischemia and 2 h of reperfusion. Polyphenol (-)-epigallocatechin gallate (EGCG) was perfused from 5 min before reperfusion to 30 min after reperfusion. Adenosine or opioid receptor antagonists were pre-treated 10 min before EGCG perfusion.
Determination of area at risk and infarct size
At the end of each experiment, the LCA perfusion circuit was precluded, and diluted fluorescent polymer microspheres (Duke Scientific Corp., Palo Alto, MA, USA) were infused to demarcate the area at risk (AR). The hearts were cut into 2-mm thick transverse slices using a rat heart slice matrix (Zivic Instruments, Pittsburgh, PA, USA). The slices were incubated in 1% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich Chemical, St. Louis, MO, USA) in sodium phosphate buffer (pH = 7.4) at 37℃ for 20 min and subsequently immersed in 10% formalin to enhance the contrast. The left ventricle (LV) was removed from the remaining tissue. The myocardial AR in the LV was identified by illuminating with UV light. The necrotic area (AN, unstained with TTC) and AR (nonfluorescent under UV light) were traced on a clear acetate transparent sheet (Fig. 2B and 2C) and quantified with UTHSCSA Image Tool, version 3.0 (Department of Dental Diagnostic Science at The University of Texas Health Science Center, San Antonio, Texas, USA). The areas were converted into volumes by multiplying them by slice thickness. The AN volume was expressed as a percentage of the AR volume. All morphometric measurements were performed in a blinded fashion by a separate technician.
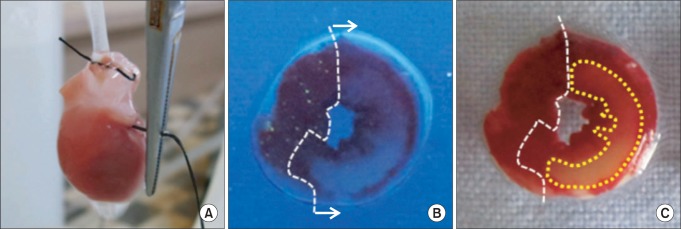
(A) Regional ischemia was induced by pulling the snare at the level of the proximal left coronary artery, (B) The area at risk of left ventricle slice was identified by UV light illumination as the tissue without fluorescence. (C) The area of necrosis was identified by unstained area by TTC (closed circle) in area at risk.
Statistical analysis
Data are presented as means ± SEM. Data analysis was performed with a personal computer statistical software package (SPSS for windows, Release 17.0; SPSS Inc, Chicago, IL, USA). Data were analyzed using one-way analysis of variance (ANOVA) with Tukey's HSD post-hoc testing. Null hypotheses of no difference were rejected if P values were less than 0.05.
Results
A total of 96 rat hearts were used for infarct measurement experiment. Four rats were excluded during the stabilization period because of a CF > 18 ml/min or < 8 ml/min (2), LVDP < 80 mmHg (1), or HR < 250 beats/min (1). A further two hearts were excluded due to irreversible post-ventricular fibrillation pump failure (1 in control and 1 in EGCG + ZM241385). Therefore, we report the data for 90 successfully completed infarct experiments (each group n = 9).
There were no significant group differences in body weight, heart weight, heart to body weight ratio, LV volume, AR volume and AR to LV ratio (Table 1). Ten µM of EGCG targeting reperfusion significantly reduced infarct size over risk area from 33.5 ± 4.1% to 14.4 ± 1.1% (P < 0.001 vs. CON) (Fig. 3). Ten µM of the nonspecific OPR antagonist naloxone (14.3 ± 1.3%, P < 0.001 vs. CON) could not block the infarct-limitation effect by EGCG. However, 1 µM of the nonspecific ADR antagonist 8-SPT (27.1 ± 1.9%, P > 0.05 vs. CON) blocked the anti-infarct effect by EGCG. Naloxone and 8-SPT itself did not alter infarct size (30.9 ± 4.5% for NAL and 29.6 ± 3.2% for 8-SPT, P > 0.05 vs. CON).

Morphometric Data (each group n = 9)
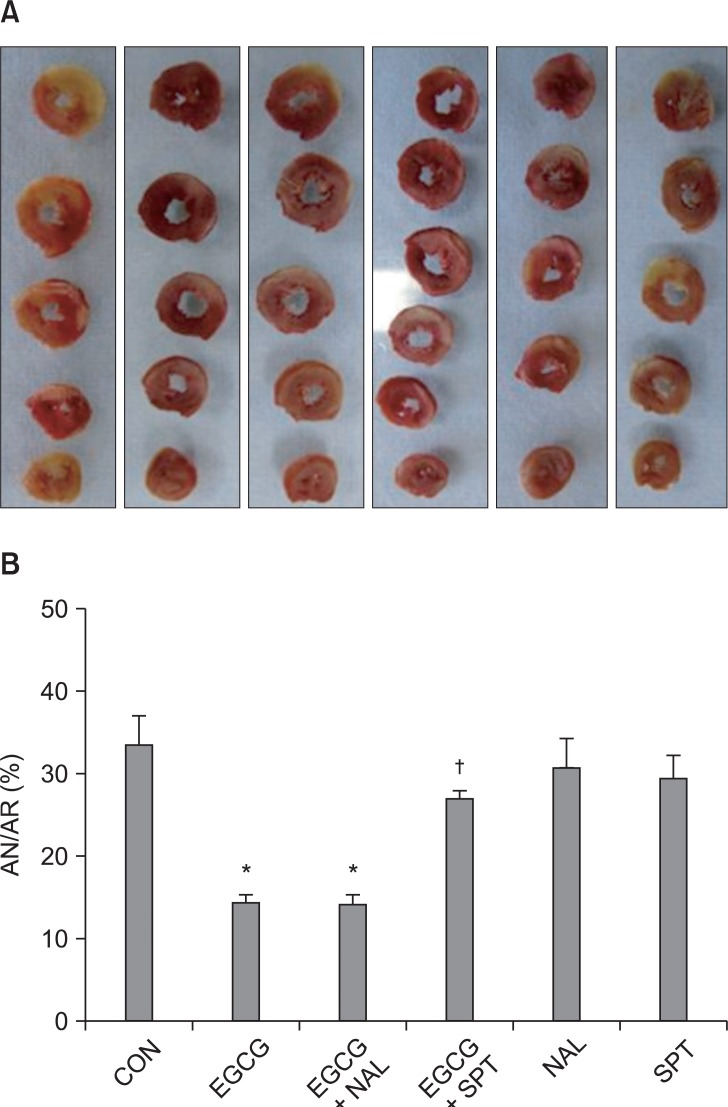
(A) Representative sequential left ventricle (LV) slices from each group showing area of necrosis (pale area) with TTC staining. (B) % of infarct area (AN) over area at risk (AR). All data are expressed as means ± SEM. CON: untreated control hearts, EGCG: polyphenol (-)-epigallocatechin gallate, NAL: nonspecific opioid receptor antagonist naloxone, SPT: nonspecific adenosine receptor antagonist 8-(p-sulfophenyl)theophylline hydrate. *P < 0.05 vs. CON, †P < 0.05 vs. EGCG.
All four ADR antagonists had a tendency to attenuate the infarct-sparing effect by EGCG. The infarct reducing effect of EGCG was significantly reversed by 200 nM of the A1 ADR antagonist DPCPX (25.9 ± 1.9%, P < 0.01) and 15 nM of the A2B ADR antagonist MRS1706 (29.3 ± 1.7%, P < 0.01) but not by 10 µM of the A2A ADR antagonist ZM241385 (23.9 ± 1.9%) and 100 nM of the A3 ADR antagonist MRS1334 (24.1 ± 1.8%) (Fig. 4).
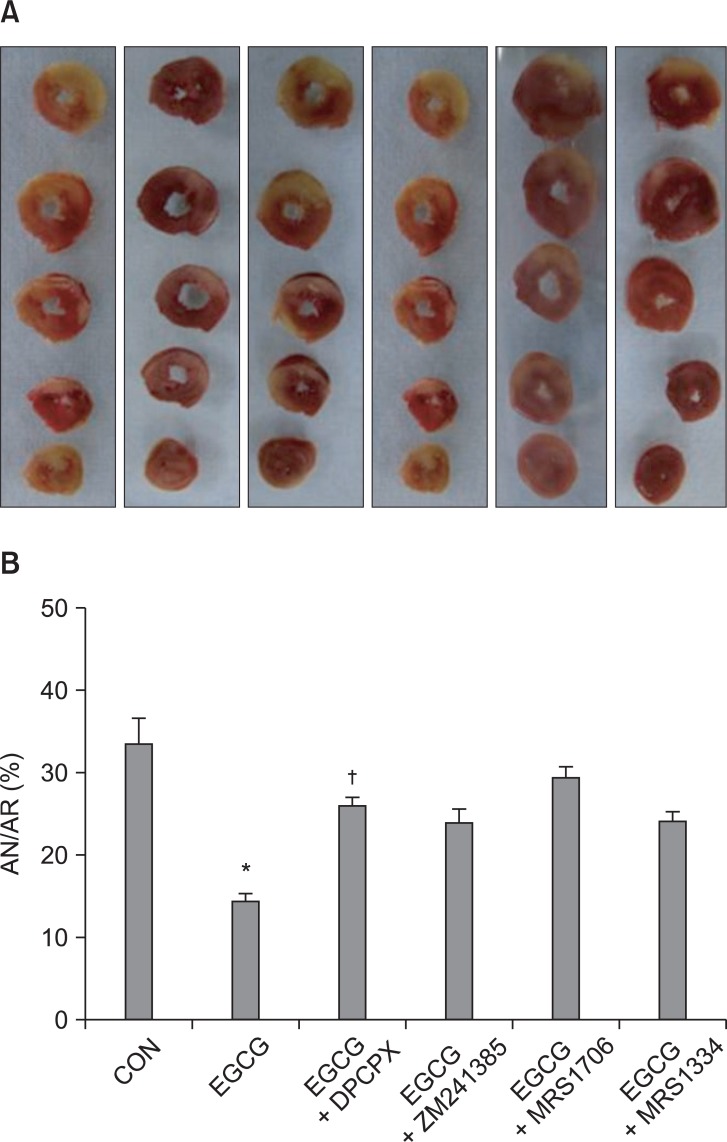
(A) Representative sequential left ventricle (LV) slices from each group showing area of necrosis (pale area) with TTC staining after adenosine receptor (ADR) antagonist pretreatment in EGCG treat hearts. (B) % of infarct area (AN) over area at risk (AR). All data are expressed as means ± SEM. CON: untreated control hearts, EGCG: polyphenol (-)-epigallocatechin gallate, DPCPX: A1 ADR antagonist, ZM241385: A2a ADR antagonist, MRS1706: A2b ADR antagonist, MRS1334: A3 ADR antagonist. *P < 0.05 vs. CON, †P < 0.05 vs. EGCG.
Discussion
In the present study, EGCG targeting reperfusion effectively reduced infarct size after myocardial ischemia and reperfusion. Interestingly, the infarct reducing effect by EGCG was totally blocked by a nonspecific ADR antagonist 8-SPT but not by a nonspecific OPR antagonist naloxone, implying the involvement of ADR in EGCG-induced cardioprotection. We further tested the involvement of subtypes of ADR in the infarct limitation effect by EGCG with four different ADR subtypes (A1, A2A, A2B, and A3) antagonists. A1 and A2B ADR subtype antagonists attenuated the infarct-sparing effect by EGCG. Our data suggest, for the first time, that EGCG-induced cardioprotection may attenuate myocardial ischemia-reperfusion injury, at least in part, through ADR activation.
Meanwhile, our present data clearly suggest that there is a functional coupling between EGCG and ADR in mediating EGCG-induced cardioprotection in the isolated rat heart, although the exact mechanism of this interaction between EGCG treatment and cardiac ADR is not known. One possible mechanism is that EGCG may further increase myocardial levels of adenosine. Adenosine is produced primarily through the metabolism of adenosine triphosphate (ATP) and its level increases during stressful situations when ATP utilization increases, such as in our myocardial ischemia reperfusion injury model. Therefore, it may be possible that EGCG further increase the release of adenosine. However, there is scant information about the adenosine level change by EGCG treatment in the heart. Navarro-Peran et al. [12] demonstrated that EGCG produces a significant increase in a specific ADR in colon cancer cells and adenosine can modulate different signaling pathways by binding to its specific receptors. In their study, A3 ADR antibody expression was significantly increased after EGCG treatment in cytosolic fractions of Caco-2 cells. Therefore, the change in adenosine level by EGCG treatment in the myocardial ischemia-reperfusion model should be determined in the future. Meanwhile, cardioprotection does not necessarily correlate with increased adenosine production. Thus, adenosine concentration is not crucial to the beneficial effects of ischemia-reperfusion of the injured rat heart [13]. In this regard, another possible mechanism might involve endogenous activation of ADR itself without increase in adenosine level.
ADR is a GPCR and there are four subtypes, A1, A2A, A2B and A3. The activation of these ADR subtypes has been shown to be cardioprotective [14-16]. During the past two decades, numerous mechanisms of adenosine-mediated cardioprotection have been proposed. ADR activation has been suggested to reduce cell death through the mitochondrial KATP (mKATP) channel as well as protein kinase C and mitogen-activated protein kinase (MAPK) signaling [17,18]. Meanwhile, we previously reported the role of mKATP channel in EGCG-induced cardioprotection in isolated rat hearts [2]. The infarct limitation effect by EGCG was totally blocked by the non-selective KATP channel blocker glibenclamide and the selective mKATP channel blocker 5-hydroxydecanoate, implying that KATP channels, especially mKATP channels, play a crucial role in the cardioprotection by EGCG. Taken together, it is highly plausible that EGCG may provide cardioprotection via mKATP channel activation by ADR activation. However, the involvement of MAPK signaling in EGCG-induced cardioprotection is not clear. Previous studies in this laboratory have shown that that EGCG treatment did not increase the phosphorylation of ERK and Akt compared to untreated control hearts (data not shown). Therefore, it is not likely that ADR activation by EGCG involves MAPK downstream signaling.
In the present study, we perfused EGCG targeting early reperfusion phase. Interestingly, it has been proposed that activation of both A1 and A3 ADR is cardioprotective when triggered ischemic period. For example, intracoronary injection of A1 ADR agonist N6-1-(phenyl-2R-isopropyl)adenosine reduced infarct size as effective as ischemic preconditioning in isolated rabbit heart [19]. In contrast, it has been demonstrated that the cardioprotection by activation of A2 ADR is beneficial during reperfusion [20,21]. Potent A2B ADR agonist NECA infused from 5 min reperfusion mimicked ischemic postconditioning's effect on infarct size in in situ rabbit hearts [22]. In this study, EGCG-induced infarct limitation was blocked by A1 and A2B ADR. This suggests ADR may be involved in the EGCG treatment targeting both ischemia and reperfusion.
In conclusion, the infarct reducing effect of EGCG appears to involve activation of ADR, especially A1 and A2B ADR, but not OPR in isolated rat heart. Further studies in an ADR knockout mouse heart model and ADR level measurement by Western blot analysis should be undertaken in the future.