The effects of hydrogen sulfide under sevoflurane administration against ischemia and reperfusion injury in isolated rat heart
Article information
Abstract
Background
Hydrogen sulfide (H2S) produces a protective effect against myocardial ischemia and reperfusion injury. Sevoflurane, which is used for anesthesia in cardiac problem patients, also has a protective effect. This study is designed to reveal the effects of H2S under sevoflurane using rat hearts.
Methods
The hearts were Langendorff-perfused, subjected to 30 minutes ischemia and 60 minutes reperfusion. Group I was a control group. The other groups were pretreated for 15 minutes before ischemia as follows: 1.6% sevoflurane for group S; 18.5 µM H2S S for group H; and 1.6% sevoflurane and 18.5 µM H2S simultaneously for group HS. Hemodynamics and the infarct size were measured.
Results
Group HS presented depressed hemodynamics during pretreatment. LV function in group HS achieved better recovery than group I after reperfusion. The infarct size of groups S, H and HS was smaller than group I, while there were no differences between groups S, H and HS.
Conclusions
Exogenous H2S did not enhance the preconditioning effects of sevoflurane. Rather, the results suggest that H2S under sevoflurane might depress hemodynamics.
Introduction
In administering anesthetics to high cardiac risk patients for surgery, the primary purpose is to maintain a cardiac reservoir. Of the many attempts to achieve this during anesthesia, a great deal of attention has been paid to the powerful defense mechanism, ischemic preconditioning that results from short repetitive myocardial ischemia. However, since there are difficulties in utilizing ischemic preconditioning during surgery [1,2], considerable research has been undertaken in the pursuit of a pharmacological solution with effects that are similar to ischemic preconditioning. Several substances, including halogenated inhalation anesthetics, have been found to cause such pharmacological preconditioning [3]. Sevoflurane is a halogenated inhalation anesthetic which has a pharmacological preconditioning effect and so is often used as a general anesthetic for patients with cardiac diseases [4].
Hydrogen sulfide (H2S) is thought to be a gasotransmitter that is naturally produced in small amounts in the human body such as nitric oxide (NO) and carbon monoxide (CO) and has many biological functions [5]. H2S is synthesized from L-cysteine through the action of cystathionine γ-lyase in the myocardium, fibroblasts and the vascular smooth muscles, and is well distributed throughout the cardiovascular system. H2S reduces the contractility of the myocardium and acts as a vasodilator leading to the dilation of vascular smooth muscles [6]. Several animal-based studies have shown that externally administered H2S has the same effect as natural H2S in reducing damage from myocardial ischemia and reperfusions [7-12]. Its myocardial protective effects are regarded as similar to that of ischemic preconditioning; H2S has a unique action in vertebrates called 'suspended animation-like state' [13]. That is, H2S induces extremely low metabolism and low oxygen consumption status such as deep hypothermia, and can protect vital organs including the heart against ischemia and reperfusion injury [13-16].
It is thought that if H2S is administrated along with sevoflurane, it can provide another effect against myocardial damage from ischemia occurring due to surgery or the course of the disease, subsequently leading to the reduction of post-operative myocardial dysfunction in high-risk patients. However, H2S is not yet in clinical use because its mechanisms, pharmacological actions and interactions with other medications are not fully understood. Therefore, this study was conducted to examine the effects of H2S on isolated rat hearts that were treated with sevoflurane and to analyze what effects the H2S would exert on the myocardium.
Materials and Methods
The study protocol was approved and all procedures were performed in accordance with the guidelines recommended by the Institutional Animal Care and Use Committees.
Our study utilized an isolated rat heart model on a Langendorff system (Radnoti Co., Colorado Springs, Colorado, USA). The hearts were taken from 52 male Spraque-Dawley white rats weighing between 250-350 g, and were divided into 4 groups by using a block randomization.
Phenobarbital 100 mg/kg was injected into the abdominal cavity of each rat and the cessation of voluntary movement was ascertained. Once the rat was completely anesthetized, 1,000 IU heparin was injected into the abdominal cavity. Before spontaneous respiration ceased, a tracheotomy was performed and a 16-G catheter was inserted and attached to an animal respirator (SN-480-7, Shinano Co., Nagano, Japan). Mechanical ventilation was initiated at 50 breaths/min, with 3-4 ml tidal volume using 95% oxygen-5% carbon dioxide. The sternum was incised and the heart was exposed. The heart was then extracted and immediately placed in iced perfusate to stop movement and prevent damage to the myocardium. The Langendorff system's perfusate cannula was inserted into the aorta of the isolated heart, with the tip positioned immediately above the aortic valve through which the perfusate was entered into the coronary artery. The perfusate used was a modified Kreb's solution (NaCl 118.0 mM, KCl 4.7 mM, CaCl2 1.9 mM, MgSO4 1.2 mM, NaHCO3 25.0 mM, KH2PO4 1.2 mM, Glucose 10.1 mM, Na-EDTA 0.5 mM). The perfusate was maintained at 37 ± 0.2℃ using a heated circulator with an automated temperature control (CW-10G, Jeio Tech Co. Seoul, Korea) and entered the Langendorff system's oxygenator through a perfusate pump (Masterflex, Cole-Parmer Instrument Co., USA). The perfusate's height was maintained at 75 cm, and the pressure of the perfusate entering the coronary artery was maintained at 55 mmHg. The perfusate's pH was maintained at 7.4 ± 0.02 by setting the oxygenator at 95% oxygen-5% carbon dioxide at 3 L/min. In order to continuously measure hemodynamic parameters, a latex balloon filled with saline was inserted through the left mitral valve into the left ventricle, and a stainless steel conduit was used to connect a pressure sensor (Ohmeda P23XL transducer, Ohmeda, Oxnard, CA, USA) to an MP-100 system (Biopac®system Inc., CA, USA) and an MP-100 manager program, Acknowledge version 3.8.2 (Biopac®system Inc., CA, USA). The other cannula was introduced through the coronary sinus via the right atrium for measuring the coronary effluent rates. Then, after stabilization, the balloon was controlled to obtain a left ventricular end diastolic pressure of around 10 mmHg before observation.
All hearts in the 4 groups were maintained on the Langendorff system, allowed to stabilize, given the initial treatment for 15 minutes, underwent artificially induced global ischemia for 30 minutes, and then underwent reperfusion for 60 minutes. Initial treatment in group I (ischemia) was simply to maintain coronary flow. Group S (sevoflurane) hearts were treated with sevoflurane at 1.6 vol% through the oxygenator using a Sevotec 5 vaporizer (Datex-Ohmeda, Tewksbury, MA, USA), group H (H2S) hearts were treated with the perfusate containing 100 µM sodium hydrosulfide corresponding to 18.5 µM H2S and group HS (H2S and sevoflurane) hearts were treated with the perfusate containing 18.5 µM H2S as well as with 1.6 vol% sevoflurane during the pre-treatment period (Fig. 1). Measurements for statistical analysis were taken directly after stabilization and after pre-treatment as well as 5, 30 and 60 minutes after initiation of reperfusion. Variables recorded for analysis were the left ventricular end systolic pressure (LVESP), left ventricular end diastolic pressure (LVEDP), maximum difference of ventricular pressure (dP/dtmax) and heart rate (HR), as well as coronary effluent flow rate (CEFR).
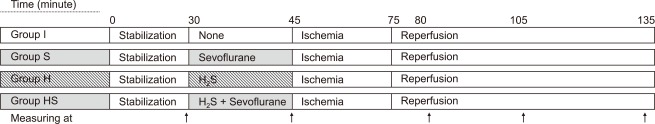
Experimental protocol. After stabilization, each group was subjected to 30 minutes of global ischemia and 60 minutes of reperfusion. Before global ischemia, a 15 minute pre-treatment period was conducted and specific agents were applied to each group as follows: perfusate and mixed gas (95% O2 + 5% CO2) only for group I; mixed gas with 1.6% sevoflurane and perfusate without H2S for group S; 18.5 µM H2S mixed perfusate and mixed gas for group H; 1.6% sevoflurane and 18.5 µM H2S for group HS. Hemodyamic variables were measured after stabilization, after pre-treatment and 5, 30, 60 minutes after reperfusion started (Arrows). After all processes were completed, triphenyl tetrazolium staining was performed to calculate infarct size.
After reperfusion was completed, the heart was immediately frozen at -20℃ to prevent damage to the muscles. All of the ventricular muscles were cut into 2 mm along the short axis of heart, and the slices were stained with triphenyl tetrazolium in a water bath of 37℃ for 10-15 minutes. The staining agent was created by dissolving triphenyl tetrazolium salt (2,3,5-Triphenyl tetrazolium chloride: C19H15ClN4) to 1% in a buffer solution of monosodium phosphate (NaH2PO4) and disodium phosphate (Na2HPO4) with a pH of 7.4. Once staining was complete, each muscle segment was fixed for 20 minutes in a 10% formaldehyde solution. The samples were then made into a uniform thickness with 2 thick glasses and 2 mm thick shims, then examined using a UTHSCSA Image Tool, version 3.0 (S. Brent Dove, University of Texas Health Science Center in San Antonio, Texas, USA). The volumes corresponded to the damaged tissues and all of the ventricles were measured and the infarct size was calculated as a ratio (%) of the sum of damaged volumes to whole ventricle.
All statistical analyses were performed using SigmaStat for Windows, version 3.0.1a (Systat software Inc., San Jose, CA, USA). Friedman repeated measures analysis of variance on ranks or Kruskal-Wallis one-way analysis of variance on ranks was used, depending on the nature of the data. When the results were significant, multiple comparisons were made with the Dunn's Method. A P value of less than 0.05 was considered statistically significant.
Results
52 rat hearts were extracted and randomly divided into 4 groups. Rats in which technical failures occurred or adequate heart function did not continue through to stabilization were excluded. In total, group I contained 10 hearts, group S contained 13, group H contained 10, and group HS contained 12.
Hemodynamic variables and CEFR
In group HS, LVESP was lower immediately after pretreatment than after stabilization (P < 0.001, Fig. 2). 5 minutes after reperfusion, LVESP was higher in group H than in groups I and S (P < 0.001). In group HS, the pressure was lower than in group H, but did not show depressed LVESP as in group S. During the remainder of the reperfusion period, the pressure decreased gradually over time. However, the pressure was higher in groups H and HS than in group I (P < 0.001).

Left ventricular end systolic pressure. Group HS showed lower systolic pressure after pre-treatment than those after stabilization. Groups H and HS showed mostly higher pressures during the reperfusion period than groups I and S. St: after stabilization, Tx: pretreatment, R5: reperfusion 5 minutes, R30: reperfusion 30 minutes, R60: reperfusion 60 minutes. P < 0.05 is remarked as follows: *vs. after stabilization in the same group. †vs. Group I, ‡vs. Group S, §vs. Group H at the same measurement time.
During the reperfusion period, LVEDP increased initially, then decreased gradually in all groups (Fig. 3). But 30 minutes after reperfusion, the pressure was lower in group S than in groups I and H (P < 0.001). Then, 60 minutes after reperfusion, the pressure was further decreased, and the differences between the groups disappeared.
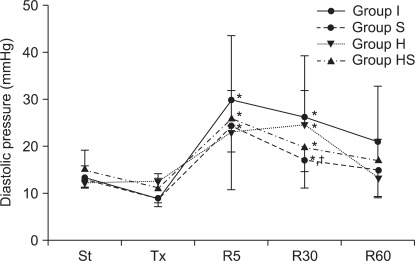
Left ventricular diastolic pressure. During the reperfusion period, diastolic pressure in all groups initially increased, and then progressively decreased over time. In group H, diastolic pressure was sustained at a high rate until 30 minutes after reperfusion and then decreased toward the value of that after stabilization. Groups S and HS showed similar patterns in pressure changes. St: after stabilization, Tx: pretreatment, R5: reperfusion 5 minutes, R30: reperfusion 30 minutes, R60: reperfusion 60 minutes. P < 0.05 is remarked as follows: *vs. after stabilization and/or pretreatment in the same group, †vs. groups I and H at the same measurement time.
All groups showed reduced HR during the reperfusion period. However, the rate was relatively higher in group S than in groups H and HS (P < 0.001, Fig. 4).
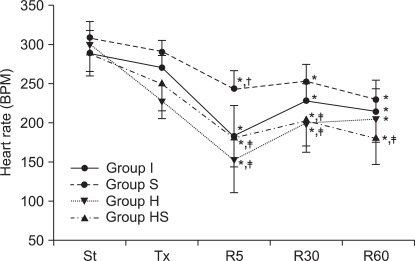
Heart rate. Heart rates during the reperfusion period in all groups were lower than those prior to ischemia. Heart rates in groups H and HS were mostly lower than those in group S during the reperfusion period. St: after stabilization, Tx : pretreatment, R5: reperfusion 5 minutes, R30: reperfusion 30 minutes, R60: reperfusion 60 minutes. P < 0.05 is remarked as follows: *vs. after stabilization in the same group, †vs. groups I, ‡vs. group S at the same measurement time.
dP/dtmax in group S abruptly decreased 5 minutes after reperfusion, similarly to group I (P < 0.001, Fig. 5). The decrement of dP/dtmax was also observed in groups H and HS, and group S showed a significantly greater drop than other groups. 30 and 60 minutes after reperfusion, dP/dtmax in all groups maintained a lowered value compared to the values measured after stabilization. In all groups, CEFR showed a gradual reduction during the reperfusion period (Fig. 6). However, in group S, the rate was generally higher than in the other groups throughout the reperfusion period (P < 0.001).
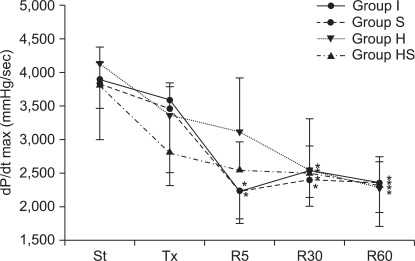
Maximum rate of change of left ventricular pressure (dP/dt max). 5 minutes after reperfusion, dP/dt max in groups I and S decreased rapidly. During the rest of reperfusion, dP/dt max of all groups remained low. St: after stabilization, Tx: pretreatment, R5: reperfusion 5 minutes, R30: reperfusion 30 minutes, R60: reperfusion 60 minutes. P < 0.05 is remarked as follows: *vs. after stabilization in the same group.
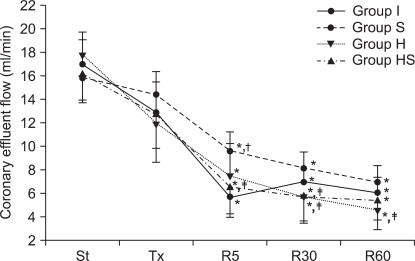
Coronoary effluent flows measured in rate. During the reperfusion period, coronary effluent flow rates of all groups decreased with time. Primarily, group S showed higher rates than groups I, H and HS. St: after stabilization, Tx: pretreatment, R5: reperfusion 5 minutes, R30: reperfusion 30 minutes, R60: reperfusion 60 minutes. P < 0.05 is remarked as follows: *vs. after stabilization in the same group. At the same measuring time, †vs. groups I, ‡vs. group S at the same measurement time.
Infarct Size (Fig. 7)
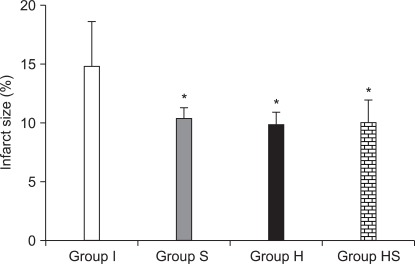
Infarct size measured in each group. Groups S, H and HS showed smaller infarct size than group I. There was no difference between infarct size of groups S, H and HS. P < 0.05 is remarked as follows: *vs. group I.
Infarct size was significantly smaller in groups S, H and HS than in group I (P = 0.001). There were no significant differences between groups S, H and HS.
Discussion
After ischemic preconditioning was introduced to prevent damage due to myocardial ischemia and reperfusion that occurs because of surgical manipulation or other causes [17], considerable research has been actively pursued and many clinical trials for the use of ischemic preconditioning are being performed [2]. Because pretreatment by short and repeated ischemia and reperfusion is highly dangerous [1], recent studies have focused on chemical pretreatments, including halogenated inhalation anesthetics and adenosine [3,4]. Among these agents, pharmacological preconditioning by inhalation anesthetics is mainly mediated through activation of protein kinase C (PKC) and ATP-sensitive potassium channels [18]. In addition, it has been demonstrated that inhalation anesthetics control the activity of nitric oxide synthase (NOS), which plays an important role in reducing ischemic damage and in maintaining CEFR [19]. Sevoflurane is a newly synthesized halogenated inhalation anesthetic and its pharmacological preconditioning effect is well known [20,21].
Our study utilized 1.6 vol% sevoflurane, which is significantly less than the 2.4%, and this corresponds to 1 MAC in rats. In our previous study, it was found that a 1.6 vol% sevoflurane, which is short of 1 MAC, had a protective effect, where a concentration higher than 1.6 vol% showed the same protective effect and instead reduced myocardial function [22]. Therefore, we chose to use 1.6 vol% sevoflurane concentration in order to prevent myocardial depression induced by sevoflurane and we found that the sevoflurane-pretreated hearts had less reduced function than group I. In addition, hearts treated with 1.6 vol% sevoflurane before ischemia had smaller infarct size compared to those in group I and so proved to have a pharmacological pretreatment effect. From a hemodynamic perspective, hearts that had been treated with sevoflurane showed a lower LVEDP, a higher HR and a higher CEFR during reperfusion than those in group I. These findings support the pharmacologic preconditioning effect of sevoflurane, as previously reported [20,21].
H2S is known to be involved in the regulation of the cardiovascular system and is accepted as a gasotransmitter, similar to NO and CO [5]. H2S was once thought to be toxic, but studies in mammals and human subjects have revealed that it is naturally produced in small amounts in the cardiovascular system. Since then, many studies have been focused on its function. In the cardiovascular system, H2S primarily functions to activate ATP-sensitive potassium channels, dilating blood vessels [6]. H2S has also been shown to suppress cytochrome c oxidase, which acts on the electron transfer system involved in cellular respiration. Thus, the suppression of oxidative phosphorylation results in extreme reduction of metabolism [13]. Moreover, externally administered H2S has an effect similar to ischemic preconditioning in reducing damage to the myocardium from ischemia and reperfusion and also in reducing infarct size. ATP-sensitive potassium channel activation appears to have some relevant effects [9,10]. Also, a previous study in rat cardiomyocytes has shown that H2S exerts a protective effect against ischemia through activation of PKC, including translocation of PKCα as well as PKCε, and PKCδ, which appears in the process of ischemic preconditioning, is slightly different in terms of PKC subtype. However, the relationships between H2S and PKC are not yet clear [11,23-25]. H2S is also believed to act as a buffer to increased oxygen consumption by regulating oxygen-consuming enzymes. In a study with mice, 150 ppm H2S gas suppressed metabolic rate by up to 90%, protecting mice from lethal hypoxia, suggesting that in hypoxia, H2S can reduce the damage to myocardial tissue [14]. Beauchamp et al. [26] have reported that high concentrations of H2S are toxic and can cause respiratory failure and pulmonary edema because it suppresses cytochrome c oxidase, disabling mitochondrial electron transport systems. It has been suggested that, through the same mechanism, low concentrations of H2S can induce reversible suspended animation-like states in mammals [13,14]. Low concentrations of H2S, in the process of metabolizing oxygen to energy in cells, lowers the oxygen demand of cytochrome c oxidase, which in turn leads to the creation of fewer reactive oxygen species (ROSs) due to sudden oxygen cutoff or hypoxia and prevents ROS-related damage to cells during ischemia [14].
The physiological concentrations of H2S in rats have been reported to vary between 10 and 50 µM, and the maximum value has been shown to be 45.6 ± 14.2 µM [6]. The concentration we used was 100 µM NaHS which corresponds to 18.5 µM H2S. This concentration was within physiological ranges and was not enough to affect the pH of the perfusate [27], and was a concentration used in previous studies [7,8,11].
We administered 18.5 µM H2S for 15 minutes prior to ischemia and found that it has a myocardial protective effect similar to sevoflurane; the infarct size in group H significantly reduced compared with group I. The reduction of infarct size was similar between the H2S and sevoflurane pretreatment. LVESP was higher in group H than in group I, HR was lower in group H than in group S, but dP/dtmax progressively decreased in later reperfusion periods. As in previous studies [9,10], H2S produced myocardial protective effects against ischemia and reperfusion injury in terms of hemodynamic aspects as well as reduced infarct size.
Our study sought to elucidate the cardioprotection of a combined treatment of inhalation anesthetics with known pharmacological preconditioning activity, and H2S which protects against myocardial ischemia and reperfusion. In the case of H2S, a mechanism similar to PKC translocation and ATP-sensitive potassium channel activation induced by inhalation anesthetics has been suggested. However, H2S induces the translocation of different PKC subtypes [11] and suppresses cytochrome c oxidase, which will lead to the suspended animation-like phenomenon which indicates a status of extremely low metabolic rate followed by a reduction in core body temperature [13,14]. Moreover, some studies cast doubt on H2S involvement in PKC translocation [25]. For these reasons, we expected good outcomes from the combined treatment with the inhalation anesthetic and H2S-induced suspended animation-like state. However, while the combined treatment reduced ischemia size, the results did not differ from those when each agent was used alone. Moreover, the combined treatment caused profound functional depression and showed no further hemodynamic benefits even though we used the minimum cardioprotective dose of each agent. In CEFR, although sevoflurane alone maintained a higher flow rate, the addition of H2S caused the rate to drop to group I levels. Generally, volatile anesthetics vasodilate coronary arteries by activation of endothelium KATP channels and promote endothelial NO release [18]. Also, H2S has a vasodilation effect; this effect is not related to KATP channels activation, and the endogenous production of NO inhibits the vasodilatory effect of H2S in coronary arteries [28]. Concerning CEFR, Geng et al. [15] reported that the effect of CEFR of H2S is dependent on the concentration of H2S. NaHS in a concentration of 100 µM did not affect the CEFR, but a higher concentration of NaHS inhibited CEFR in addition to a severe negative inotropic effect. These findings suggest that combined treatment with inhalation anesthetic and H2S could have an adverse effect on CEFR and our experimental results support this effect.
However, it cannot be assumed that this completely rules out any possible additional benefits from the combined treatment. First, further studies are needed to ascertain whether H2S-induced metabolic suppression leads to temporary myocardial dysfunction. In order to determine this, we should examine metabolism in the control myocardium, each agent separately, as well as in combined treatment. Additionally, in in vivo experiments, we should look for early preconditioning effects after 60 minutes of reperfusion and late preconditioning effects 2-3 days after ischemia. In our research, combined treatment with sevoflurane and H2S led to no additional benefit compared to a single treatment with sevoflurane or H2S as an early preconditioning effect. Second, there is a possibility that a rate-limiting factor and a ceiling effect of cardioprotection exists in pharmacological preconditioning. Sevoflurane-induced cardioprotection against ischemia and reperfusion continues to increase as concentrations increase, but only up to 1.6% [22]. NaHS administered between the preischemic state and late reperfusion state in doses of 0.1 µM and 1 µM showed a progressive decrease in ischemia size, but at 10 µM this effect did not occur [9]. Based on these results, including those of our study, more research is needed to determine what happens in the interaction between sevoflurane and naturally occurring H2S, considering its role as a gasotransmitter and a pharmacological preconditioner that reduces ischemia size.
In conclusion, we observed that H2S administered to isolated rat hearts had preconditioning effects against ischemia and reperfusion injury, such as faster systolic pressure recovery and reduction of infarct size. Sevoflurane was found to have preconditioning effects, as reported previously. Combination treatment with H2S and sevoflurane has an effect on infarct size reduction but caused a greater loss of hemodynamic function than a single treatment with H2S or sevoflurane for each substance.