Introduction
Among nerve agents, soman is a major threat partly because of the serial toxicologic events following exposure to this nerve agent. It is a potent irreversible acetylcholinesterase (AChE) inhibitor in the cholinergic nervous system, which can produce serial toxic effects [1]. Reactive oxygen species (ROS) derived neuronal injury is one of the main mechanisms of delayed neuronal injury caused by soman. Intoxication with sublethal to lethal doses of soman causes convulsive seizures and seizure-related brain damage which are mediated by ROS production.
ROS derived from hydrogen peroxide (H2O2), superoxide (O2), and peroxynitrite (ONOO) are highly reactive and can lead to cell injury [2]. The injured cells exhibit serious responses such as inflammation and programmed cell death (e.g. apoptosis). Apoptosis is explained by controlled auto-degradation of cells and plays important roles in many biological processes including cellular damage, tumorigenesis, and teratogenicity [3]. Alternatively, to protect cells from damage induced by oxidative stress, cells have anti-oxidant defense systems and survival-promoting pathways. The rapid activation or induction of protective enzymes which decrease oxidative stress by reducing ROS increases the capacity of cells to maintain homeostasis during oxidative stress; activation of survival-promoting pathways allow cells to tolerate and/or to recover from the damage [4]. Therefore, modulating these anti-oxidative defense systems and survival pathways may influence ROS derived cell injury.
Midazolam, a benzodiazepine derivative, is the most widely used anesthetic for sedation. It is also used in critically ill patients who usually suffer from the pathologic effects of oxidative stress such as infection, hemodynamic instability and hypoxia [5,6]. Several lines of evidences support an interaction between midazolam and ROS. Midazolam interferes with the synthesis and release of nitric oxide and tumor necrosis factor-╬▒ (TNF-╬▒) generated by activated immune cells [7] and inhibits neutrophil apoptosis in the perioperative periods. This suggests that midazolam may modulate inflammatory cellular injury.
Through studies of the anti-inflammatory effects of midazolam, we hoped to support the idea that midazolam can protect the Central Nervous System (CNS) from soman intoxication. However, it was necessary to first determine whether midazolam activates cell protective mechanisms that are involved in soman derived neural cell injury. Therefore, the aim of the present study was to explore the hypothesis that midazolam protects B35 neuroblastoma cells from ROS stress through activation of survival-promoting pathways such as anti-apoptotic signaling.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin, streptomycin, trypsin/EDTA and glutamine were supplied by Gibco-BRL (Rockville, MD, USA). ECL western blotting detection reagents and PRO-PREP protein extract solution were supplied by iNtRON Biotechnology (Houston, TX, USA). Anti-p-Akt antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-╬▓-actin antibody, Glucose oxidase (GOX) and other chemicals were purchased from Sigma (St. Louis, MO, USA).
Cell culture
B35 neuroblastoma cells were purchased from American Type Culture Collection (Manassas, VA, USA). For all experiments, cells were grown in DMEM supplemented with 100 U/ml penicillin, 0.1 mg/ml streptomycin and 10% heat inactivated FBS in culture plates. The cells were incubated in a 37Ōäā incubator under 5% CO2 in a humidified atmosphere. After reaching confluence, cells were pretreated with various drugs depending on the experimental design.
Detection of LDH activity
Cell viability changes due to GOX and culture conditions were determined in terms of LDH released into media of cell cultures. Cells in the exponential phase of growth were seeded at 1 ├Ś 104 cells/well in 24-well plates. After different treatments, 50 ml of standards or 50 ml of culture supernatant (controls and treated cells) and 50 ml of substrate mix were transferred into a 96-well-plate. After incubation for 30 min at room temperature, plates were read using a SUNRISE ELISA-plate Reader (Tecan, Crailsheim, Germany) at 490 nm. LDH released from cells is expressed as a percentage of total cellular LDH.
Western blot analysis
Whole cell lysates were isolated using PRO-PREP protein extract solution. The whole cell lysates were centrifuged at 100,000 g for 20 min at 4Ōäā. The protein concentration in each lysate was determined by the Bradford assay. To detect Akt-phosphorylation, 30 micrograms of protein were subjected to 10% SDS-polyacrylamide gel electrophoresis for 90 min at 120 V depending on the molecular size of the phospho-Akt protein. The separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes for 2 h at 20 mA using SD Semidry Transfer Cells (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were stained with Ponceau S solution to determine efficiency of transfer and/or protein loading levels. Then the PVDF membranes were blocked for 2 h at room temperature with 5% nonfat milk in Tris-buffed saline (TBS, pH 7.0) containing 0.05% Tween 20 (TBS-T). The membranes were incubated with an anti-phospho-Akt antibody overnight at 4Ōäā, using a dilution of 1 : 500 in 5% bovine serum albumin (BSA) in TBS-T. For the detection of bound antibody, horseradish peroxidase conjugated anti-goat or anti-rabbit IgG was used. The signals were detected by ECL using a western blotting luminol reagent system (iNtRON Biotechnology, Houston, TX, USA) and autoradiography.
FACS analysis
A total of 1 ├Ś 107 cells were seeded in 100 mm culture dishes, incubated at 37Ōäā, and harvested at the indicated times after mitosis. Cells were stained with propidium iodide (PI) using a previously described method [8]. The harvested cells were trypsinized, washed twice in PBS, and fixed in 70% ethanol on ice for a minimum of 2 h. Cells were washed 2 additional times with PBS and stained for 30 min at 37Ōäā in 50 ┬Ąg/ml PI solution containing 200 ┬Ąg/ml RNase A and 0.1% Triton X-100. After incubation of samples in the dark for 30 minutes, cell cycle profiles were determined using a Beckton-Dickinson flow cytometer (Becton Dickenson Biosciences, San Jose, CA, USA) with CellQuest┬« Software.
Results
Akt phosphorylation is responsible for the protective effect of midazolam against ROS damage
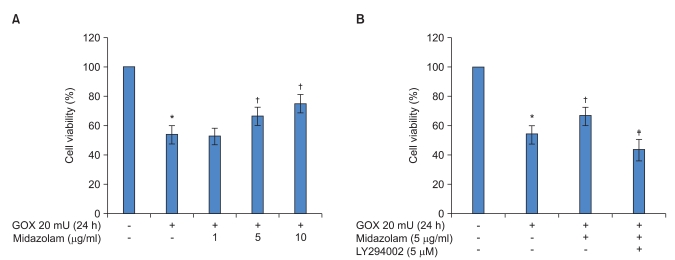
The effects of midazolam on cell viability during ROS injury were analyzed by LDH assay. As expected, 10 h of incubation with 20 mU/ml of GOX significantly decreased cell viability. Fig. 1A shows that GOX derived ROS injury decreased cell viability about 1.6-2 times compared to the control. However, treatment with midazolam (5 and 10 ┬Ąg/ml) concentration-dependently reduced the decreased cell viability due to ROS injury, and this difference was statistically significant. To determine the effect of an Akt-phosphorylation inhibitor on cell viability, 5 ┬ĄM concentrations of LY294002 were used to treat cells for 1 h prior to treatment with midazolam. As shown in Fig. 1B, LY294002 inhibited the protective effect of midazolam. For example, pretreatment with 5 ┬ĄM LY294002 significantly decreased cell viability that had been induced by midazolam.
Dose-dependent induction of Akt-phosphorylation by midazolam is mediated through PI3 kinase activation
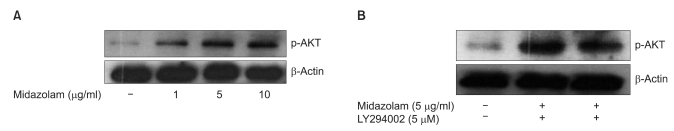
We next asked whether midazolam can protect B35 cells from oxidative stress induced injury by regulating Akt-phosphorylation. Akt phosphorylation plays important roles in general cellular defense systems against oxidative stress in mammalian cells. As shown in Fig. 2, western blot analysis indicated that midazolam-induced cytoprotective effects in ROS injury may be mediated by induction of Akt-phosphorylation through PI3 kinase activation. The effect of midazolam on Akt-phosphorylation was dose dependent, but the induction was abolished by pretreatment with LY294002, a PI3 kinase inhibitor.
Midazolam inhibits GOX-induced apoptosis through Akt phosphorylation
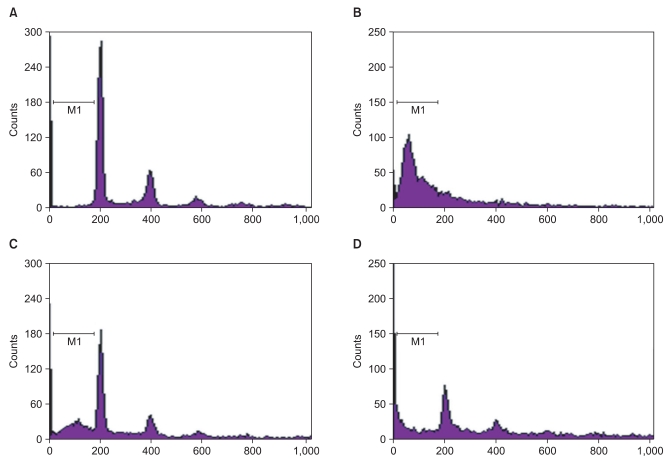
It is generally known that ROS induces apoptosis in many mammalian cells [9] and GOX is frequently used as a chronic ROS producing agent [10]. Therefore, we investigated whether midazolam protects cells from GOX-induced apoptosis. Fig. 3 shows that GOX-induced apoptosis was diminished by midazolam pretreatment. The effect is mediated through Akt-phosphorylation because the diminished apoptosis due to addition of midazolam became obvious again as the concentration of LY294002, an inhibitor of PI3 kinase, increased. FACS analysis (Fig. 3) showed that about 61% of cells underwent apoptosis after GOX (20 U/ml), but pretreatment with midazolam significantly reduced the fraction of dead cells to 21%. Co-treatment with LY294002 significantly reduced the anti-apoptotic effect of midazolam.
Discussion
The major finding of this study is that the midazolam increases Akt phosphorylation through PI3 kinase activation and significantly decreases GOX-induced apoptotic cell death in B35 cells. This conclusion is based on the findings that : (1) midazolam concentration-dependently induces phosphorylation of Akt in B35 cells; (2) LY-294002, a PI3 kinase inhibitor, significantly inhibits the protective effect of midazolam against ROS injury.
For application of the results of the present study to an in vivo study, pharmacological concentrations have to be considered. The concentrations used (1, 5 and 10 ┬Ąg/ml) in the present study were chosen based on the report of Hudson [11]. In the report, midazolam was given to patients with abdominal aortic surgery at 0.25 mg/kg and the maximum plasma concentration (Cmax) ranged from 200 to 3,000 ng/ml. Several studies reported that midazolam has antioxidant effects, and these effects could make midazolam effective against pathologic states mediated by oxidative stresses [5,6]. Interestingly, the findings in this study indicate that the beneficial effects of midazolam on ROS-induced pathologic states may also be regulated by induction of protective mechanisms such as Akt-phosphorylation.
However, the finding in the present study does not coincide with the study of Stevens et al. [12]. They reported pro-apoptotic effects of midazolam in human Jurkat T-lymphoma cells and human neuroblastoma cells via regulation of mitochondrial pathways at midazolam concentrations ranging from 100 to 400 ┬ĄM (over 24 hr). These finding are not consistent with results of the present study, but this could be because several factors in the two studies were different. We exposed cells for only 8 hrs. The concentrations of midazolam and the duration of the pre-incubation step were different. In the present study, we used midazolam concentrations of 1, 5 and 10 ┬Ąg/ml which correspond, respectively, to 0.31 ┬ĄM (1 ┬Ąg/ml), 1.53 ┬ĄM (5 ┬Ąg/ml) and 3.1 ┬ĄM (10 ┬Ąg/ml). Thus, midazolam may induce apoptosis at higher concentrations that are more toxic concentration but this has to be evaluated further to confirm that midazolam has dual effects on apoptosis depending on its concentration.
Generally, it is well-recognized that Akt-phosphorylation plays a major role in cell proliferation and survival in many cell types and is classically activated by phosphoinositide-dependent kinases following recruitment to the plasma membrane by products of the type I phosphoinositide 3-kinase [13]. However, the fact that midazolam may serve a critical role in a cell's protective mechanism during oxidative stress through Akt phosphorylation is a novel finding.
Originally, this exploratory study was done to determine whether midazolam can protect cells from soman induced neural injury. Because ROS are involved in the neurotoxicity or neuronal injury during soman intoxication [14,15], we deliberately used 20 mU/ml GOX to simulate a pathologic state where the generation of sustained and continual release of H2O2 can give rise to oxidative stress [16]. Therefore, the antiapoptotic effect of midazolam in GOX-induced ROS injury supports the idea that midazolam has a beneficial effect in soman intoxication.
In summary, we showed that midazolam, a benzodiazepine, leads to phosphorylation of Akt in neuronal cells through activation of the PI3 kinase pathway. This induction of Akt-phosphorylation is responsible for the protection of B35 cells from GOX-induced oxidative stress. Thus, we conclude that midazolam is important in the management of soman-induced neuronal injury.