The optimal concentration of siRNA for gene silencing in primary cultured astrocytes and microglial cells of rats
Article information
Abstract
Background
Small interfering RNAs (siRNAs) have been used to knockdown specific gene expression in various cells. Astrocytes and microglial cells play a key role in fundamental central nervous system functions and in chronic neuroinflammation. The aims of this study were to determine the optimal concentration of siRNA demonstrating efficient transfection and inhibition of gene expression via RNA interference (RNAi) and lower cytotoxicity, in primary cultured astrocytes and microglial cells of rats.
Methods
Astrocytes and microglial cells were isolated from the cerebral cortices of 2-day-old rats. Both the cells were transfected using transfection reagent (Lipofectamine™ 2000), and fluorescein-labeled double-stranded RNA (dsRNA) or siRNA targeting green fluorescent protein. Transfection efficiency and cytotoxicity of dsRNA, and the degrees of RNAi induced by siRNA in these cells, were evaluated at various concentrations of RNA.
Results
Transfection efficiencies of dsRNA in both astrocytes and microglial cells were significantly higher (P < 0.05) at the concentrations of 20, 40, and 80 nM than at the concentrations of 0, 5, and 10 nM. There were no significant cytotoxicities within the applied concentrations of dsRNA (0-80 nM). The degrees of RNAi induced by siRNA were significantly higher (P < 0.05) at the concentrations of 5, 10, 20, 40, 80 nM, and 20, 40, 80 nM in astrocytes and microglial cells, respectively, compared with the control (0 nM).
Conclusions
The siRNA concentration of 20 nM may be appropriate to induce RNAi in both astrocytes and microglial cells, while demonstrating low cytotoxicity, high transfection efficiency, and effective RNAi.
Introduction
RNA interference (RNAi) is a naturally occurring mechanism for regulating gene expression which has been observed in several models of organisms, and is mediated by double stranded RNA (dsRNA) [1-3]. The intracellular presence of dsRNA homologous to a gene results in post-transcriptional gene silencing via induced sequence-specific degradation of the corresponding mRNA. In recent pain research, it has been demonstrated that astrocytes and microglial cells release a variety of proinflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor-α, which contribute to the pathogenesis of neuropathic pain [4-7]. Silencing of gene expression in glial cells may be an alternative treatment approach for neuropathic pain. In practice, small interfering RNAs (siRNAs) have been applied successfully to down regulate gene expression of neuronal cells in the central nervous system [8].
However, RNAi may result in an artificial dysregulation of non-target genes, which are called off-target effects [9]. Off-target effects can be mainly explained by two mechanisms. One is induction of the interferon response in mammalian cells after transfection of RNA molecules, and the other is unintended targeting of genes that have only low level of sequence homology to the RNA molecule [9,10]. In general, unwanted effects, such as activation of the interferon response, are more likely to occur at high siRNA concentrations [9,11,12]. siRNA concentrations of less than 20 nM usually do not lead to induction of the interferon response [11], although an earlier study observed off-target effects at the concentration of siRNA of 10 nM [12]. For the effective and safe application of siRNA in astrocytes and microglial cells, it is necessary to establish appropriate siRNA concentrations for efficient transfection into these cells and silencing of target gene without producing off-target effects and cytotoxicity.
The aims of this study were to determine the optimal concentrations of siRNA demonstrating efficient transfection and inhibition of gene expression via RNAi and lower cytotoxicity, in primary cultured astrocytes and microglial cells of rats.
Materials and Methods
Materials
Insulin, transferrin, putrescine, thyroxine, triiodothyronine, progesterone, selenium, soybean trypsin inhibitor, bovine pancreatic DNase, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich, Inc. (St. Louis, MO, USA). Collangenase was purchased from Worthington Biochemical Corp. (Lakewood, NJ, USA). Glutamine, 20% BSA soultion, Dulbecco's modified Eagle's medium (DMEM), trypsin-EDTA, L-15 medium, fluorescein-labeled dsRNA oligmer (BLOCK-iT™ fluorescent oligo), transfection reagent (Lipofectamine™ 2000), and fetal bovine serum (FBS) were purchased from Invitrogen Corp. (Carlsbad, CA, USA).
Preparation of siRNAs
The siRNA sequence targeting plasmid encoding green fluorescent protein (pEGFP-C1) was selected from a previous study and modified [13]. The sequences of anti-green fluorescent protein (GFP) siRNAs used were 5'-GCA CGA CUU CUU CAA GUC CGC CAdT dG-3' (active anti-GFP siRNA). The active anti-GFP siRNA was synthesized from Samchully Pharm. Co., Ltd. (Siheung, Korea).
Primary culture of astrocytes and microglial cells
Two-day-old neonatal Sprague Dawley rats were obtained from Orient Bio Co., Ltd. (Seongnam, Korea). The number of neonates which were used in one primary culture was 12-15 heads from one pregnant rat, and five primary cultures were conducted in this study. All procedures were approved (No. 2006-01-059) by the Institutional Animal Care and Use Committee of the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea. The Committee abides by the dictates of the Institute of Laboratory Animal Resources guide [14]. Cultures were prepared from the whole brains of 2-day-old Sprague Dawley rats using the procedures described in previous studies [15,16]. Cortex obtained from neonatal rats, was placed in Hanks' balanced salt solution, and then meningeal tissues, hippocampus, caudate nucleus, and other internal structures were removed to leave only the cortical sheets. The cortical tissues were minced gently with pipette and mixed with collagenase, and then were incubated for 30 minutes at 37℃. After centrifuging at 1,000 rpm for 5 minutes, the supernatants were removed and the tissues were resuspended in trypsin-EDTA solution. After incubation for 5 minutes at 37℃, soybean trypsin inhibitor and DNase solution were added for 5 minutes. After centrifuging at 1,000 rpm for 5 minutes, the supernatants were removed, and modified-DMEM was added and tissues were triturated into a cell suspension using 5-ml pipette and 18-gauge needle. Cells were filtered through a mesh bag (40 µm), mixed with DMEM containing FBS 10%, penicillin 100 U/ml, and streptomycin 100 mg/ml, and then were plated on 75 cm2 culture flasks and kept in DMEM supplemented with FBS 10% and the antibiotics in a humidified 5% carbon dioxide atmosphere at 37℃, and the medium was changed every 2 days. After 12-13 days, mixed glial cultures were separated into astrocytes and microglial cells.
Mixed glial cultures grown for 12 or 13 days in 75 cm2 flasks were shaken on a rotary platform at 150 rpm for 2 hours at 37℃. After shaking, the supernatants were used for harvesting the microglial cells, and the remaining source cultures were dissociated using trypsin-EDTA, and then collected by centrifugation (1,000 rpm for 5 minutes). The cells were seeded onto culture plates of each experiment and were cultured for 24 hours before being used as astrocyte cultures. Harvested supernatants were seeded onto culture plates of each experiment, and then were incubated for 10 minutes at 37℃. Non-adherent or weakly adherent cells were removed by gentle shaking and washed out. The remaining cells were cultured for 24 hours and used as microglial cultures.
Immunocytochemistry
The astrocytes and microglial cells were seeded onto poly-D-lysine coated coverslips (BD BioCoat, BD Biosciences, San Jose, CA, USA) at 2 × 104 cells/cm2 and 4 × 104 cells/cm2, respectively, and cultured for 24 hours before staining. The cultures were fixed with paraformaldehyde 4% in phosphate-buffered saline (PBS) 0.1 M for 10 minutes and rinsed three times with PBS 0.1 M. Cells were incubated with 0.25% Triton X-100 for 10 minutes at room temperature. Non-specific binding was blocked with 10% BSA for 30 minutes at room temperature. Subsequently, cultures were incubated with mouse monoclonal antibodies (1 : 200 dilution) to the specific markers of microglia (OX-42 against CD 11b surface antigen, ABR, Golden, CO, USA) or astrocytes (glial fibrillary acidic protein [GFAP], Sigma-Aldrich, Inc., St. Louis, MO, USA) in the blocking buffer for 24 hours at 4℃. After washing three times with PBS, the cells were incubated with the secondary antibody (Alexa Fluor 488 goat anti-mouse IgG for GFAP, Alexa Fluor 594 goat anti-mouse IgG for OX-42, 1 : 1,000 dilution, Molecular Probes, Eugene, OR, USA) for 2 hours at room temperature and rinsed. The preparations were mounted in fluorescent mounting medium (Dako, Glostrup, Denmark) and examined using a confocal fluorescence microscope (Leica TCS SP2, Leica microsystems, Bensheim, Germany). We calculated the percentage of positively immunostained cells (in 200 cells) in each of the three separate culture preparations.
siRNA uptake assessment
The primary astrocytes were seeded at a density of 4 × 104 cells/well and microglial cells were seeded at a density of 1 × 105 cells/well in 24-well plate, and then grown for 24 hours prior to transfection. Transfection efficiencies of dsRNA in astrocytes and microglial cells were tested using fluorescein-labeled dsRNA oligmer (the BLOCK-iT™ Fluorescent Oligo), which has the same length, charge and configuration as the standard siRNA, and is strictly designed for use as a tool for siRNA uptake assessment. Fluorescein-labeled dsRNA oligmer were prepared in the concentrations of 0, 5, 10, 20, 40 and 80 nM, and mixed with 1 µl of transfection reagent. The complex of dsRNA and transfection reagent was transfected into both the cells using Opti-MEM I (Invitrogen Corp.) without serum. This medium was replaced with growth medium at 4-6 hours after transfection.
Evaluation of siRNA effects using fluorescence detection of GFP
The primary astrocytes were seeded at a density of 1 × 105 cells/well and microglial cells were seeded at a density of 4 × 105 cells/well in 6-well plate, and then grown for 24 hours prior to transfection. Anti-GFP siRNA was prepared in the concentrations of 0, 5, 10, 20, 40 and 80 nM, and mixed with transfection reagent (2.5 µl) and pEGFP-C1 (800 ng). The complex was transfected into both the cells using Opti-MEM I without serum. This medium was replaced with growth medium at 4-6 hours after transfection.
Evaluation of siRNA effects using reverse transcription polymerase chain reaction
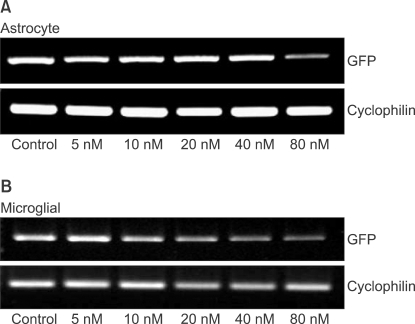
The levels of GFP gene products were determined by reverse transcription polymerase chain reaction (RT-PCR) as described previously [13]. The RNA (500 ng) prepared from the transfected cells was reverse-transcribed into first-strand cDNA with oligo (dT)15 primer (PrimeScript 1st Strand cDNA Synthesis Kit, Takara Korea Biomedical Inc., Seoul, Korea) in 20 µl reactions. The PCR was performed with 1 µl of the first-strand cDNA solution and 0.5 µM each of the sense and antisense primers using PCR kit (PrimeScript 1st Strand cDNA Synthesis Kit, Takara Korea Biomedical Inc., Seoul, Korea) for 30 cycles of denaturation (94℃, 30 seconds), annealing (62℃, 30 seconds) and extension (72℃, 30 seconds); GFP, or for 30 cycles of denaturation (94℃, 30 seconds), annealing (59℃, 30 seconds) and extension (72℃, 30 seconds); cyclophilin. Primers and PCR product sizes for the corresponding target genes were as follows: GFP (5'-3': forward, AAC GGC CAC AAG TTC AGC GTG T; reverse, ACA GCT CGT CCA TGC CGA GAG T ; base pair, 642), cyclophilin (5'-3': forward, ATG TGC CAG GGT GGT GAC TTC A; reverse, TTG TCC ACA GTC GGA GAT GGT; base pair, 379). The PCR products were electrophorated on 1.5% agarose gels, stained with ethidium bromide and visualized by ultraviolet (UV) illumination. Digital images of the PCR products were produced under an UV light. The intensity of PCR product staining, which has been shown to be directly proportional to the amount of DNA, was determined using a densitometer (VersaDoc Multi-Imaging Analyzer Systems, Bio-Rad, Hercules, CA, USA). The amount of each PCR product of GFP was normalized to cyclophilin levels, a transcript whose levels do not change in contused spinal cord [17].
Image analysis
To evaluate transfection efficiency and modulation effect for GFP gene expression by siRNA, fluorescent uptake was assessed at 16 hours post-transfection using confocal fluorescence microscope (standard FITC filter set; λex = 494 nm, λem = 519 nm green). Images from the confocal microscope were stored on hard disk (512 × 512 pixel format) or erasable optical disk. The nuclear and total cellular fluorescence in each confocal slice were determined using image software (ImageJ; NIH image, http://rsbweb.nih.gov/nih-image). For each complex, we randomly selected two areas per well, and then the transfection efficiency of each trasfection reagent and fluorescein-labeled dsRNA oligmer complex, and modulation effect for GFP gene expression by siRNA were expressed as a percentage of the transfected areas (astrocyte, measured fluorescent positive area/total measured area) and transfected cells (microglia, counted fluorescent positive cell/total counted cell).
Cytotoxicity of dsRNA
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, Inc., St. Louis) reduction activity was measured with a colorimetric assay. The primary astrocytes were seeded at a density of 5 × 104 cells/well and microglial cells were seeded at a density of 5 × 105 cells/well in 96-well plate, and then grown for 24 hours prior to transfection. At 4 hours after transfection with transfection reagent (0.25 µl) and dsRNA, which was prepared in the concentrations of 0, 5, 10, 20, 40 and 80 nM, the mixture was replaced with 0.1 ml of fresh medium. The cells were incubated for an additional 24 hours at 37℃. Ten L of MTT dye solution (5 mg/ml in phosphate buffer pH 7.4) was added to each well. After 2 hours of incubation at 37℃ and 5% CO2 for exponentially growing cells and 15 minutes for steady-state confluent cells, the medium was removed and formazan crystals were solubilized with 100 µl of 0.04 N-HCl in isopropyl alcohol, and the solution was vigorously mixed to dissolve the reacted dye. The absorbance of each well was read on a microplate reader (Spectramax 340PC, MDC, Sunnyvale, CA, USA) at 570 nm. The spectrophotometer was calibrated to zero absorbance, using culture medium without cells. The cells that were exposed to 10 µM ionomycin (Sigma-Aldrich, Inc.) for 24 hours were used 100% cell death (full-kill) [18]. Cell viability was calculated as follows:
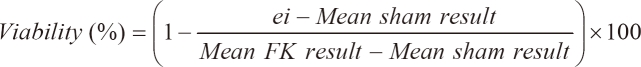
where ei refers to the absorbance of each well containing each dsRNA concentrations. Mean sham result is the mean absorbance of five replicated 0 nM concentration. Mean FK result is obtained by averaging the results from each of the wells treated with a substance that produces 100% cell death conditions. Tests were performed as five replicated experiments of the three experiments (n = 15).
Statistical analysis
The experimental results were expressed as mean ± SD. The cytoxicity, transfection efficiency and GFP gene expression levels between the concentrations of dsRNA or siRNA were compared using Kruskal-Wallis test, and then pairwise multiple comparisons were performed using the Mann-Whitney U-test. The Mann-Whitney U-test was used to compare the mRNA expression levels between the different concentrations of anti-GFP siRNAs. A value of P < 0.05 was considered statistically significant. The statistical analyses were performed with SPSS (version 12.0, SPSS Inc., Chicago, IL, USA).
Results
Purity of primary cultured cells
Astrocytes and microglial cells from mixed glial cultures were isolated to identify the cell types in which siRNA acts. The purities of the astrocytes and microglial cultures were 96% and 94%, respectively, as determined by immunocytochemistry (Fig. 1).
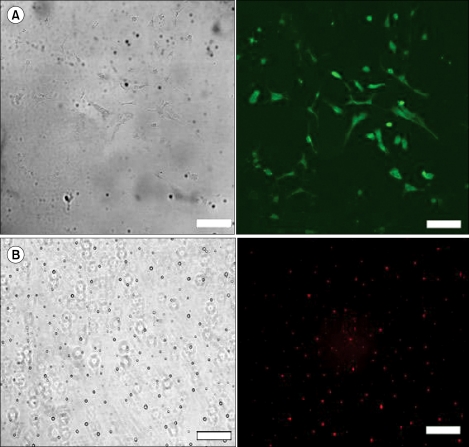
Representat ive photomicrographs of secondary cultures of both astrocytes (A) and microglial cells (B) subjected to immunocytochemical analysis. Left panels show phase-contrast micrographs and right panels show fluorescence micrographs. The majority of cells in astrocyte cultures were positively stained with an antibody to glial fibrillary acidic protein. The majority of cells in microglial cultures were immunostained with OX-42 (×200, scale bar = 100 µm. OX-42 = specific markers of microglia [antibody against CD 11b surface antigen]).
Transfection efficiency of dsRNA in primary cultured astrocytes and microglial cells
With increase in fluorescent-labeled dsRNA concentration, transfection efficiencies of dsRNA on primary cultured astrocytes and microglial cells increased. Transfection efficiencies of dsRNA in both astrocytes and microglial cells were significantly higher (P < 0.05) at the concentrations of 20, 40, and 80 nM than at the concentrations of 0, 5, and 10 nM (Fig. 2).
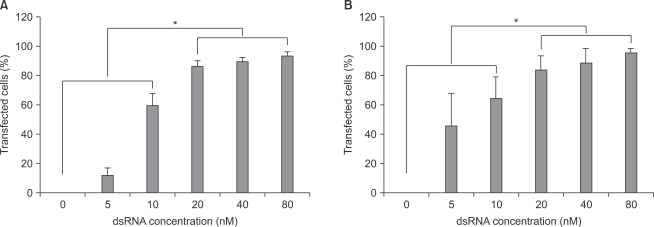
Transfection efficiency of dsRNA in both primary cultured astrocytes (A) and microglial (B) cells. The transfection efficiency was observed by confocal fluorescence microscope (standard fluorescein isothiocyanate filter set; λex = 494 nm, λem = 519 nm green) and the number of transfected cells was analyzed by ImageJ program (NIH image, http://rsbweb.nih.gov/nih-image). The transfected cells values are means ± SD of three independent experiments (*P < 0.05, n = 6). dsRNA: fluorescein-labeled dsRNA oligmer.
Cytotoxicity of dsRNA in astrocytes and microglial cells
As shown in Fig. 3, there was no significant cytotoxicity within the applied concentrations of fluorescein-labeled dsRNA in both astrocytes and microglial cells.
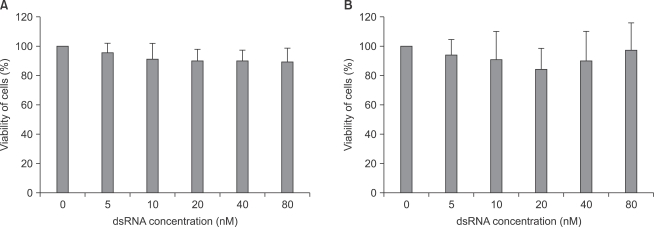
The cytotoxicity of dsRNA in both astrocytes (A) and microglial cells (B). Cells were transfected with transfection reagent and dsRNA complex, which were prepared in concentrations of 0, 5, 10, 20, 40 and 80 nM of dsRNA, and incubated for 4 hours and an additional 24 hours at 37℃. The cellular viability was measured by MTT assay. The percent viability of cells are means ± SD of three independent experiments (n = 15). dsRNA: fluorescein-labeled dsRNA oligmer.
Modulation of GFP gene expression by siRNA
As shown in image analyses of fluorescence (Fig. 4 and 5), silencing effects induced by anti-GFP siRNA on the expression of GFP gene were significantly higher (P < 0.05) at the concentrations of 5, 10, 20, 40, 80 nM, and 20, 40, 80 nM in astrocytes and microglial cells, respectively, compared with the control (0 nM). The expression levels of GFP mRNA evaluated using RT-PCR in both astrocytes and microglial cells were consistent with image analyses for fluorescence (Fig. 6).
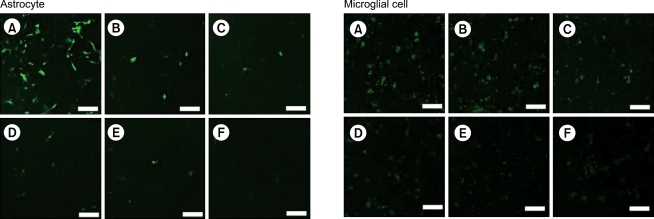
The degree of RNAi increased with increasing concentration of anti-GFP siRNA in astrocytes (upper panels) and microglial cells (lower panels). Cells were transfected with plasmid encoding GFP (pEGFP-C1) of 800 ng and each concentration of anti-GFP siRNA (A: 0 nM, B: 5 nM, C: 10 nM, D: 20 nM, E: 40 nM, and F: 80 nM). The fluorescence of GFP was observed under confocal fluorescence microscope (×200, scale bar = 100 µm). RNAi: RNA interference, GFP: green fluorescent protein, siRNA: small interfering RNAs.
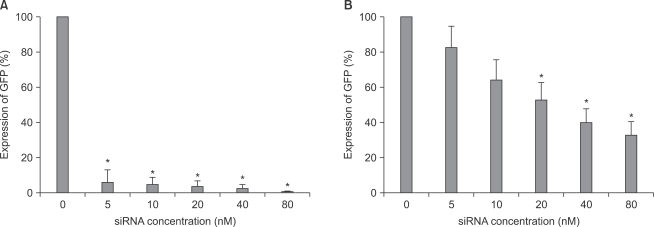
The quantification of GFP gene expression using image analysis in both astrocytes (A) and microglial cells (B). The GFP gene expression level was determined by counting the number of transfected cells (counted fluorescent positive cell/total counted cell). The transfected cells values are means ± SD of three independent experiments (*P < 0.05, n = 6). GFP: green fluorescent protein.
The quantification of GFP gene expression using image analysis in both astrocytes (A) and microglial cells (B). The GFP gene expression level was determined by counting the number of transfected cells (counted fluorescent positive cell/total counted cell). The transfected cells values are means ± SD of three independent experiments (*P < 0.05, n = 6). GFP: green fluorescent protein.
Discussion
High transfection efficiency of siRNA-lipopolymer complexes is of crucial importance for gene silencing in transfected cells. Transfection efficiency of fluorescein-labeled dsRNA was determined in HeLa and P19 cells, which was observed to be high in the earlier studies [19,20]. In this study, astrocytes and microglial cells also showed high transfection efficiencies (>80%) of fluorescein-labeled dsRNA at the concentrations of 20 nM or higher, which were concentration-dependent within these ranges of dsRNA concentrations (20-80 nM) as well.
There have been controversies regarding the concentration of transfected dsRNA molecules to prevent off-target effects. In earlier studies, these concentrations were known to be 5, 10, or 20 nM [11,12,21]. This study demonstrated that the lowest in vitro concentrations of anti-GFP siRNA silencing a target gene were different between astrocytes (5 nM) and microglial cells (20 nM). In addition, fluorescein-labeled dsRNA showed poor transfection efficiencies at the concentrations of less than 20 nM. Therefore, the lowest concentrations of siRNA for silencing target mRNAs in both the cells should be about 20 nM, considering the in vivo conditions where mixed astrocytes and microglial cells exist. Off-target effects of siRNA were first screened by microarray profiling experiments [11,21-23], and then confirmed by evaluating the expression of specific genes such as CYLD and SOAT gene in siRNA targeting GFP [21]. The types of off-target effects associated with siRNAs were currently recognized as follows: microRNA-like off-target effects [9,22], immune stimulation [12], and saturation of the RNAi machinery [24]. Hence, understanding the sources of off-target activity is yielding insights into the techniques to mitigate them.
The MTT assay for cell viability evaluation has been described as a suitable method for detection of biomaterial toxicity, and this assay relies on the mitochondrial activity of cultured cells and represents a parameter for their metabolic activity [25]. The mitochondrial metabolism of astrocytes and microglial cells was assumed to be stable at the concentrations of RNA applied in this study.
Since siRNA molecules cannot cross the blood-brain barrier, direct site injections into the central nervous system, such as intrathecal administration, are required under in vivo conditions [26]. In the field of pain research, typically, 1-2 g of siRNA daily are administered intrathecally into rats weighing between 200-220 g, in order to induce gene silencing [26,27]. In addition, the volume of injectate into the intrathecal space of rats should not exceed 10% of the total cerebrospinal fluid (CSF) volume (approximately 250 L) to avoid CSF hypervolemia and to minimize costs associated with transfection reagents [26].
In conclusion, the lowest concentration of dsRNA required for efficient transfection into astrocytes and microglial cells was 20 nM. The dsRNA concentrations of 80 nM or lower did not cause cytotoxic effects in both the cells. In addition, the lowest concentrations of siRNA required for inhibiting the expression of targeted gene (GFP) in astrocytes and microglial cells were 5 nM and 20 nM, respectively. Therefore, the siRNA concentration of 20 nM may be appropriate to induce RNAi in primary cultured astrocytes and microglial cells of rats, while demonstrating low cytotoxicity, high transfection efficiency, and effective RNAi.
Notes
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (No. 2009-0088527).