Introduction
An injured peripheral nerve can be the cause of abnormalities of the autonomic, sensory or motor nervous system at the damaged area, and the neuropathic pain may encompass functional disability. The number of patients with neuropathic pain conditions, including cancer pain, postherpetic neuralgia, diabetic neuropathy, and complex regional pain syndrome (CRPS) shows an increasing trend [1,2]. Studies on the psychosocial impact of neuropathic pain indicate that it reduces quality of life, affecting mood as well as physical and social functioning [3]. The causes and mechanisms of neuropathic pain have yet to be clarified, and efforts to ease the pain by means of opioid analgesics, antidepressants, and anticonvulsants have been marginally effective. According to the research on the development of neuropathic pain, excitatory amino acids (EAAs), which increase in levels in the spine after spinal nerve damage, activate many pain-related receptors and increase the concentration of calcium within cells. EAAs acting at the N-methyl-D-aspartate (NMDA) receptor in particular, contribute to central sensitization and persistent nociception following tissue injury. The activation of NMDA receptors increases the amount of intracellular MAPKs, such as p38, c-Jun NH2-terminal kinase (JNK), and extracellular signal-regulated kinase 1/2 (ERK 1/2), as well as the calmodulin kinase (CaMK) through adenylyl cyclase, protein kinase A (PKA), and protein kinase C (PKC) [4-7]. In addition, damaged peripheral nerves induce inflammatory responses considered to be the determining cause of the neuropathic pain [8]. Thus, this study was devised upon the idea that the use of ketamine, the antagonist of the NMDA receptor, will reduce both the production of p38 MAPK and the inflammation, and suppress the development of the neuropathic pain.
Materials and Methods
This study was conducted under the permission and rules of the Institutional Laboratory Animal Care and the Ethical Committee of the University. For the research, male Sprague-Dawley rats weighing between 180-200 g were used, following the Kim and Chung method [9] for the preparation and behavioral analysis of the neuropathic pain models. The operation was performed after anesthetizing the animals by injecting pentobarbital sodium (50 mg/kg i.p. or to effect). An incision was made from the 3rd lumbar vertebra to the 2nd sacral vertebra through the mid-line of the vertebrae, followed by removal of 6th transverse process of the lumbar vertebra by pulling the muscles on the left side, in order to expose the 5th and 6th lumbar spinal nerves. These nerves were tied with 5-0 silk suture to induce the nervous damage before the muscles and skin were sutured closed. A week after the damage was initiated, the withdrawal threshold of the models' paws was analyzed using von Frey filaments (0.6, 1.0, 2.0, 4.0, 6.0, 8.0, 10. 0, 15.0 g; Stoelting Co., Wood Dale, IL, USA) as the mechanical stimulation. The rats that demonstrated the withdrawal reaction under 4 g of bending force were considered the successful neuropathic pain model group [10]. Rats that underwent an identical operation procedure, but without tied nerves, were considered the sham-operated group. Rats that exhibited motor deficiencies or those that failed to exhibit subsequent mechanical allodynia were excluded from the experiment. In order to observe the effect of ketamine on the neuropathic pain, the neuropathic and ketamine group (NP + Keta group, n = 8), which had ketamine infused into the subcutaneous tissue using implanted mini-osmotic pumps (model 2ML1, DURECT Corporation, Cupertino, CA, USA) at the speed of 10 ┬Ąl/h (40 mg/kg/day) for 7 d [11], and the neuropathic pain and normal saline group (NP + NS group, n = 8), with normal saline infused instead of ketamine were compared to the sham-operated group. The mechanical withdrawal threshold test was carried out every day for 7 d following the implantation of the mini-osmotic pumps. At day 7 following pump implantation, the rats were deeply anesthetized with oxygen and 5% sevoflurane. The rats were then decapitated and their brains were harvested. The sacrum and 3rd lumbar vertebra were cut almost simultaneously, and the spinal cords were harvested by rapidly infusing physiologic saline into the spinal canals and pushing the spinal cords toward the lumbar vertebrae. The harvested brain and spinal cord tissues were quickly frozen using liquid nitrogen and stored in the -80Ōäā refrigerator. The changes in phospho-p38 MAPK and p38 MAPK signaling proteins were detected through immunoblotting analyses as described previously [12]. The mRNA expression of the pro-inflammatory cytokines, tumor necrosis factor ╬▒ (TNF╬▒), and intercellular adhesion molecule 1 (ICAM1) was assessed by reverse transcriptase-polymerase chain reaction (RT-PCR) [13,14]. All examinations and statistical analyses were carried out double-blinded with respect to the treatment groups of the animals.
Western blot analyses for phospho-p38 and p38 MAPK
The refrigerated brains and spinal cords of the rats were dissected on ice, and were instantly soaked in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, and 1% Triton-X100 [pH 7.4]) for 10 s to be homogenized. The tissue was ultra-centrifuged (32,500 rpm, 4Ōäā, vacuum) for 1 h and the upper liquid layer was collected. The amount of protein in the liquid was determined using the Bradford assay (BioRad, PA, USA) with bovine serum albumin as the standard, and the protein was stabilized with Laemmli buffer. Each protein sample (50 ┬Ąg) was electrophoresed for 4 h at 80 V through a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel, and the protein were then transferred to polyvinylidene fluoride (PVDF) membrane through the transfer unit at 20 V, 4Ōäā, overnight. The PVDF membrane was washed thoroughly using Tris-Buffered Saline Tween-20 (TBST, 0.1% Tween 20 in 1 ├Ś TBS) solution and coated with blocking solution (5% dry milk in TBST). CST antibody (Cell Signaling Technology, Inc., Danvers, MA, USA) was used as the primary antibody for phospho-p38 and total-p38, and after the PVDF membrane was washed with TBST solution, it was incubated with the secondary antibody (goat anti-rabbit or anti-mouse IgG) for 1 h and then developed using electrochemiluminescence (ECL) reagents (Amersham Biosciences Corp., Piscataway, NJ, USA) and ultraviolet products, with Bio-Imaging System (UVP LLC, Upland, CA, USA).
Semi-quantitative reverse transcriptase polymerase chain reaction (RT-PCR) for mRNAs, TNF╬▒ and ICAM1
The brain and spinal cord tissues of rats were placed in 1 ml of Trizol reagent (Molecular Research Center, Inc., Cincinnati, OH, USA), and were homogenized before being allowed to react at room temperature for 5 min. Then, 0.2 ml of 1-bromo-3-chloropropane was added (Sigma-Aldrich, St. Louis, MO, USA) and centrifugation was performed for 15 min at 14,000 g. Subsequently, after separating the supernatants and adding an equal amount of phenol: chloroform (5: 1; pH 4.7) (Sigma-Aldrich), centrifugation was carried out for 10 min at 14,000 g. 100% ethanol was added to the separated supernatants, which were then allowed to react for 1 h at -20Ōäā and then centrifuged for 10 min at 14,000 g. The upper liquid layer was removed and the RNA precipitate was washed with 75% ethanol, after which diethylenepyrocarbonate (DEPC) water was added and, the RNA was allowed to react for 5 min at 60Ōäā and then refrigerated. The concentrations of the extracted RNA were determined by spectrophotometric absorbance at 260 nm. With extracted RNA (0.5 ┬Ąg) as the template, cDNA (50 ┬Ąl) was composed using avian myeloblastosis virus (AMV) reverse transcriptase (Promega, Madison, WI, USA). PCR was performed in a reaction volume of 10 ┬Ąl, including dNTPs (each at 2.5 mM/L), 1 unit I Start Taq DNA polymerase (Intron Biotechnology, Seoul, Korea), and 20 ┬ĄM/L of each primer, with 2 ul of the composed cDNA (Table 1). PCR was carried out through 35 repeated cycles of 30 s of denaturation at 95Ōäā, 30 s of annealing at 55Ōäā, and 1 min of extension at 72Ōäā, and after the cycles, 10 min of reaction at 72Ōäā. A ╬▓-actin of 228 bp was used as the internal control gene to confirm the separation of appropriate mRNA from cells. The extended PCR products were electrophoresed in a 2.0% agarose gel, dyed with ethidium bromide, and quantification analysis was performed using Gel DocŌäó XR (Bio-Rad Laboratories, Philadelphia, PA, USA).
Statistics
All values were represented as mean ┬▒ SEM with SPSS 12.0 (SPSS Inc., Chicago, IL, USA) used for statistical processing. A comparison of mechanical withdrawal threshold between two groups was done with Student's t-test. In addition, oneway ANOVA was used for comparison of western blots and RT-PCRs among the three groups, and if a significant difference was found, Tukey's post hoc test was used. A P value lower than 0.05, was considered statistically significant.
Results
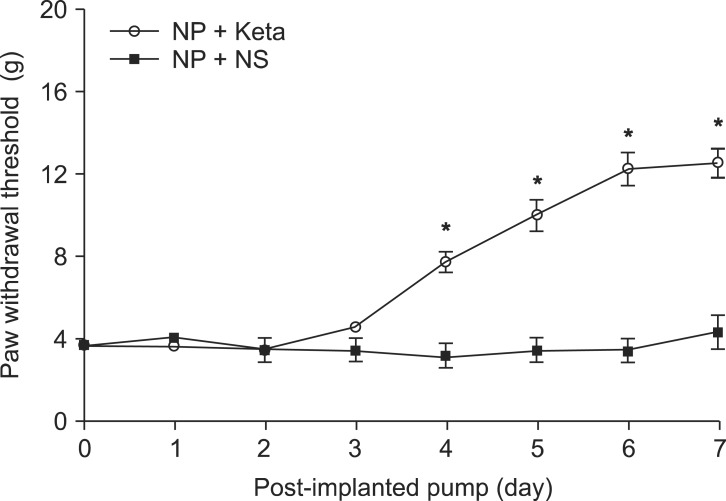
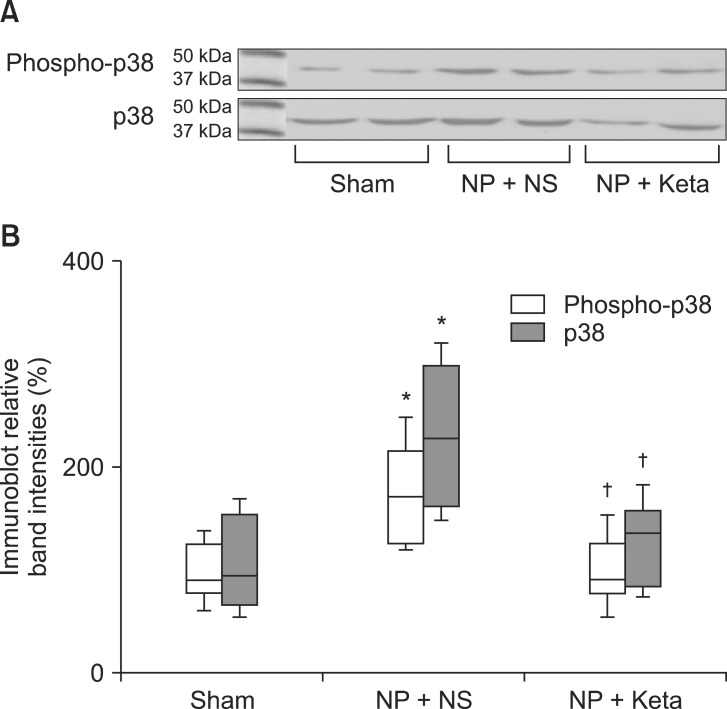
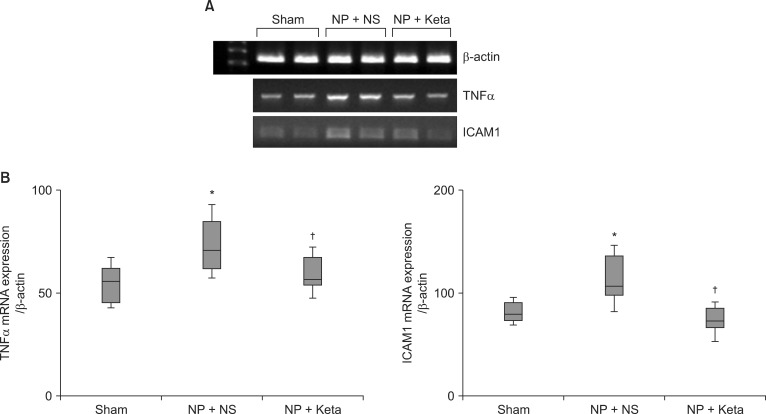
Among neuropathic rats confirmed through behavioral testing that received implanted mini-osmotic pumps and were observed for one week with a paw withdrawal threshold test, the NP + Keta group showed notable recovery of withdrawal threshold after the 4th day of testing. However, the NP + NS group maintained its threshold without a significant difference (Fig. 1). The western blot analysis using brain tissues and the mRNA expression comparison after RT-PCR of TNF╬▒ and ICAM1 showed no significant differences among the control group, the NP + NS group, and the NP + Keta group. However, the quantitative changes of phospho-p38 MAPK and total p38 MAPK observed in the spinal cord tissues were seen to be 178.4 ┬▒ 17.9 and 238.4 ┬▒ 26.8 respectively, in the NP + NS group. These values represented increases of 76 and 127% (P = 0.003, 0.001, respectively) from the control group values of 101.4 ┬▒ 10.4 and 104.9 ┬▒ 16.4. In contrast, the NP + Keta group showed significant decreases (P = 0.004, 0.005, respectively) of 75 and 93% from the values of the NP + NS group, to 102.1 ┬▒ 12.3 and 123.7 ┬▒ 14.8 (Fig. 2). There were no significant differences in signaling protein changes between the NP + Keta group and the control group (Fig. 2). The RT-PCR analysis on TNF╬▒ and ICAM1 in the spinal nerves showed a noticeable increase in the mRNA expression of TNF╬▒ and ICAM1 in the NP + NS group compared to the control group (P = 0.008, 0.006, respectively). On the other hand, the NP + Keta group had its mRNA expression of TNF╬▒ and, ICAM1 significantly decreased in comparison to the NP + NS group (P = 0.02, 0.002, respectively), but showed no significant changes in comparison to the control group (Fig. 3).
Discussion
In this study, we confirmed that the p38 MAPK and proinflammatory cytokines increased markedly in a rat model of neuropathic pain, and the use of ketamine in neuropathic rats significantly decreased not only the increased p38 MAPK, but also the inflammatory responses. In addition, we observed the elevated mechanical threshold of neuropathic symptoms through behavioral testing in the group that received ketamine.
The development of neuropathic pain contributes to the production of several neuropathic symptoms that express diverse individual pathophysiologic mechanisms in the peripheral and central nervous systems [15]. The generally known pathology of neuropathic pain is the histological and biological distortion in the spinal dorsal horn by the sensitization of central nerves that leads to continuous hyperexcitability in processing of afferent nociceptive pathways. Tissue damage, inflammation, or injury of the nervous system may result in chronic neuropathic pain characterized by hyperalgesia, allodynia, and spontaneous pain [16]. Excitatory amino acids, such as glutamate and aspartate, play an important role in the ischemic cascade leading to cell death. The NMDA receptor is an excitatory amino acid (EAA) receptor, and NMDA receptor antagonists have been shown to exert a neuroprotective effect in central nervous system ischemia [17]. Furthermore, elevated EEAs lead to the activation of several MAPKs, including ERK 1/2, JNK, and p38 [18,19]. The key element in this process is the glutamate receptor that increases in number at neural junctions by phosphorylation, elevating the reactivity to glutamate, an excitatory neurotransmitter [20]. The recent advance in molecular biology give stronger impetus to expanding the scope of the study of intracellular signaling pathways. The NMDA receptor, a G protein-coupled receptor (GPCR), is a transmembrane receptor located on the neural network. It is one of the ionotropic glutamate receptors (ligand-gated ion channels), along with AMPA and kainate. When an ionotropic receptor is activated, a channel that allows the flow of Na+, K+, and Cl- is opened. While the activation of a metabotropic receptor also opens the ion channel through a number of reactions, it must be accompanied by secondary neuropathic elements (cAMP, cGMP, IP3, DAG, etc.) [21]. Numerous receptors related to GPCRs interfere with the action of ion channels, and therefore influence the action of PKC, the glutamate transporter (EAAT3), phosphatidylinositol 3-kinase (PI3K), MAPKs, CREB, and many others that are intracellular signaling proteins [22]. According to Labombarda et al. [23], in consideration of the observed increase in PKC and NMDA receptors after damage to spinal nerves, the quantitative increase and activation of the NMDA receptor is critical in developing and prolonging neuropathic pain.
Moon et al. [24] showed that direct activation of spinal sigma-1 receptors induces mechanical allodynia with phosphorylation of p38 MAPK's involved in the process. In addition, activation of the p38 MAPK pathway through transient receptor potential A1 in the dorsal root ganglion contributes to the development and maintenance of inflammatory and neuropathic pain. Garry et al. [25] reported that increased p38 and p42/44 (ERK 1/2) MAPK in the spinal cord after spinal constriction injury of mice are important in the development of chronic neuropathic pain, with cytokines, vasoactive intestinal peptide receptor (VPAC2), neurokinin (NK2), produced in a neuroglial cell and NMDA receptor during the process are related. As seen, most the of research on the mechanisms of neuropathic pain notes the relationship between the significant quantity of MAPKs, including ERK 1/2 and p38, and the appearance of CREB protein within the intracellular signaling pathway through GPCRs. The activation of the NMDA receptor at the spinal cord level increases the concentration of Ca2+ within cells, activating nitric oxide and PKC. The phosphorylation of NO-mediated protein kinases and PKC strengthens the receptor and Ca2+ pathway complex at the cell membrane, eventually resulting in neuropathic pain. In addition, PKC and NO-mediated protein kinase made by the activation of the NMDA receptor decrease the efficiency of the mu opioid receptor and the processes it controls, as well as interfering with the coupling of the mu opioid receptor and G proteins, resulting in increased tolerance to morphine's analgesic effect [26]. According to Wagley et al. [27], the close relationship between the expression of the mu opioid receptor, JNK, and the p38 MAPK pathway makes it possible to use MAPK inhibitors to counter tolerance to morphine and reduce withdrawal symptoms. This research aims to clarify the cause of neuropathic pain through the quantitative analysis of p38 MAPK and pro-inflammatory cytokines among the cell signaling proteins in a rat model of neuropathic pain, and the effect of the use of ketamine, the antagonist of the NMDA receptor, on p38 MAPK and antiinflammatory responses. Ketamine, besides blocking activated ion channels by combining with the junction site of phencyclidine, weakens the harmful noxious afferent stimulus as a noncompetitive antagonist to EAAs, and precludes the wind-up effect in the spinal dorsal horn, resulting in suppression of mechanical allodynia and hyperalgesia, the characteristics that led to its occasional use in neuropathic pain patients [28]. Ketamine's antagonistic effect at the NMDA receptor resulted in decreased activation of neurons related to tissue damage along with decreased reaction to noxious stimuli, which in turn led to reduction in the symptoms of neuropathic pain. There are reports of other effects of ketamine related to pain control, such as that related to its similarity to the local anesthetics. Schnoebel et al. [29] reported that ketamine is concentration-dependent, reversibly suppresses the conductance of Na+ channels in human nerve cells, and alters the voltage dependent activity and inactivity of the ion channels. More recently, a clinical trial was conducted on the analgesic and antiinflammatory effects of losmapimod, an inhibitor of p38 MAPK, as a treatment for rheumatic arthritis [30]. This current research was carried out under the expectation that reduction of p38 MAPK that is related to the cell signaling of GPCRs, as well as reduction of the inflammation, an important causative factor in neuropathic pain, similar to the effect achieved with local anesthetics, will be possible through the repetitive use of small doses of ketamine to suppress activation of the NMDA receptor at the spinal cord level.
In conclusion, we observed a significant increase of the intracellular signaling protein, p38 MAPK and the proinflammatory cytokines, TNF╬▒ and ICAM1 in a rat model of neuropathic pain induced by spinal nerve ligation. These findings, inversely, show that the MAPKs, including p38, and inflammatory responses contribute to the development of neuropathic pain. It is thought that the small doses of ketamine used as treatments for neuropathic pain suppressed the activity of GPCRs by interfering with the function of G╬▒ protein and controlling the flow of ions through the Ca2+ or K+ channels, which decreased the production of p38 MAPK signaling protein. In addition, the use of ketamine reduces inflammation, which is also thought to be an important cause of neuropathic pain, prevents the development of neuropathic pain, and can attenuate the acute tolerance induced by the opioids that are commonly used in cancer pain patients, possibly resulting in decreases in both the dosages used and the adverse effects of opioids in these patients.