Pheochromocytoma (PHEO) is a neuroectodermal tumor that originates in the chromaffin cells of the sympathetic nervous system. It occurs in approximately 0.002% of the general population [1]. In 90% of cases, the PHEO is located in the adrenal medulla. The remaining 10% are extra-adrenal or retroperitoneal PHEO (Paraganglioma [PGL]), originating in the carotid body, Organ of Zuckerkandl, and other paraganglia of the autonomic nervous system [2]. Almost all PGLs (75%) are located near the aorta in the Organ of Zuckerkandl, an extra-adrenal chromaffin tissue near the origin of the inferior mesenteric artery system. PGLs most frequently occur in patients in their 20s and 30s, and are slightly more common in males. This is in contrast to adrenal PHEOs, which are typically diagnosed in patients in their 40s and 50s, and are slightly more common in females [3]. PGL has various clinical manifestations, attributable to the excessive secretion of catecholamines by the tumor. The most common symptoms are paroxysmal hypertension, headache, palpitations, and sweating. Infrequently, atypical symptoms such as nausea, vomiting, and epigastric discomfort may appear. Atypical clinical presentations, or even cases that are completely asymptomatic, can make diagnosis difficult at times. Extraluminal perigastric masses may be frequently presumed to be gastrointestinal stromal tumors (GISTs) in the absence of histologic confirmation. Therefore, in cases in which typical symptoms of catecholamine excess are not present, and a histologic diagnosis is lacking, perigastric PGLs may be mistaken for GISTs. As a result, any patients presenting for anesthesia could potentially be harboring an undiagnosed PGL, with likely disastrous consequences.
Case Report
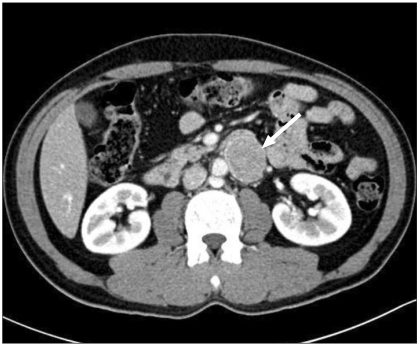
A 44-year-old male patient was admitted to the hospital for surgical removal of a duodenal submucosal tumor found incidentally on duodenoscopy. An abdominal CT scan showed a well-defined heterogeneous retroperitoneal enhancing mass measuring 3.6 ├Ś 4.5 ├Ś 3.1 cm in the paraaortic space (Fig. 1). There were no distant abdominal metastatic lesions. The patient was prepared for surgery for suspected GIST with a duodenal origin. The patient's height was 170 cm and weight 68 kg. He had a 20-year history of smoking and was on medication for hyperlipidemia, but had no other significant past medical or family history. Blood tests were normal except for an elevated amylase of 242 IU/L.
The patient was not premedicated. Prior to anesthetic induction, the patient's blood pressure (BP) was 130/75 mmHg and his heart rate (HR) was 74/min. The modified Allen test was positive and a 20-gauge catheter was placed into the radial artery for continuous BP monitoring. Induction of anesthesia was done with propofol 150 mg followed by vecuronium 10 mg. Desflurane 8 vol% and remifentanil 0.2 ┬Ąg/kg/min were continued for 3 minutes prior to endotracheal intubation. Desflurane 4 vol%, N2O 50%, and remifentanil 0.06 ┬Ąg/kg/min were used for maintenance of anesthesia. The patient remained hemodynamically stable during induction, intubation, and surgical incision. Bispectral index was maintained at 35-50. During the initial manipulation of the tumor, at the beginning of the surgery, the patient became hypertensive, with a BP of 250/120 mmHg, and tachycardic, with a HR of 100/min. The hypertension was treated with a combination of sodium nitroprusside infusion (0.5 ┬Ąg/kg/min), labetalol hydrochloride (10 mg of labetalol in two doses of 5 mg each), remifentanil infusion (0.3 ┬Ąg/kg/min), and increased desflurane concentration up to 9 vol%. Although the BP decreased to 155/100 mmHg following the infusion, there was an increase in the systolic BP (above 180 mmHg) and the HR (95-105/min) each time the surgeon manipulated the tumor. Despite the adequate depth of anesthesia and anti-hypertensive therapy, the BP remained difficult to control. Furthermore, the electrocardiography (ECG) showed T wave inversion. The surgeon was asked to stop the tumor manipulation. Subsequently, an additional dose of nicardipine hydrochloride 300 ┬Ąg was administered, and isosorbide dinitrate 0.2 ┬Ąg/kg/min infusion was maintained. After the administration of nicardipine, the BP initially decreased to 130/80 mmHg, and systolic BP was maintained at 140-150 mmHg thereafter. An arterial blood gas analysis performed at this point showed pH 7.35, PaCO2 40 mmHg, PaO2 168 mmHg, and oxygen saturation 99% on FiO2 0.5. Cardiac enzymes were drawn. Twenty minutes after the surgery was halted, the ECG returned to normal. The surgery was therefore resumed. Desflurane 9 vol%, remifentanil 0.3 ┬Ąg/kg/min, sodium nitroprusside (0.1-0.3 ┬Ąg/kg/min) and isosorbide dinitrate 0.2 ┬Ąg/kg/min were administered continuously, and the BP remained stable at around 130-160 mmHg systolic. There were no changes in the ECG after the resumption of surgery, and the troponin I levels checked afterwards remained below 0.20 ng/ml. As the BP gradually decreased with ligation of the tumor, desflurane and remifentanil were tapered. The sodium nitroprusside infusion was discontinued upon tumor ligation. One hundred and fifty minutes after the start of the surgery, the tumor was completely removed. Following tumor removal, the BP decreased to as low as 85/48 mmHg and the HR increased 110/min. Estimated blood loss during the surgical procedure was about 500 ml. In order to improve the patient's hemodynamic parameters, volume expanders (500 ml) and crystalloid fluids (1,000 ml) were given, and 8 mg of ephedrine was administered intravenously. After these procedures, the BP returned to 140/87 mmHg and the HR returned to 88/min. The patient's vital signs were maintained with desflurane 4 vol% and remifentanil 0.03 ┬Ąg/kg/min until the surgery was completed. The patient was then extubated and moved to the intensive care unit.
An undiagnosed extra-adrenal PHEO was suspected based on the hypertensive crisis caused by tumor manipulation during the surgery. A biopsy showed that the tumor was a well-encapsulated, solid mass measuring 5.5 ├Ś 4.0 ├Ś 3.7 cm. Capsular invasion was seen, but neither lymphovascular invasion nor necrosis was found. Characteristics of neuroendocrine carcinoma were seen with hematoxylin and eosin staining. Immunohistochemical staining revealed the neuroendocrine markers synaptophysin and chromogranin, and a positive CD56 confirmed the diagnosis of PGL. The patient was transferred to the general ward on postoperative day (POD) 2 and was discharged on POD 11 without any complications. A follow-up appointment was scheduled for six months after the surgery, to investigate any evidence of recurrence and to monitor signs of lymph node metastasis.
Discussion
Extra-adrenal PHEO, or PGL, shows clinical signs of catecholamine excess. The most common catecholamine secreted is norepinephrine, and the classic triad of symptoms of catecholamine excess is headache, palpitations, and sweating [4]. Sustained or paroxysmal hypertension has been reported in 80-100% of patients. The main manifestation of sympathetic PGL is hypertension, and this type of tumor is usually localized in the thorax or the abdomen. While parasympathetic PGLs are mostly localized around the head and neck, these tumors are nonfunctional, and biochemically silent [5]. There are cases, therefore, in which the classic triad of symptoms does not occur. In addition, some cases are either asymptomatic or ambiguous, with confounding factors including psychiatric disorders, anxiety, facial pallor, weight loss, polyuria, hyperglycemia, secondary erythrocytosis, stroke, and cardiomyopathy, making a proper diagnosis difficult [4]. In such cases, the patient may be unaware that he or she has PGL until it is discovered incidentally from an imaging study such as a CT or MRI. However, specific patient interviewing could help trigger the suspicion of PGL. There is also a special need for a physical examination to determine the presence of cardiovascular side effects from such a tumor. In hindsight, the patient described here showed a borderline BP elevation before surgery; therefore, a physician should not overlook borderline elevation of the BP in patients. Also, PGL or PHEO should be considered in patients with a history of resolved Takotsubo cardiomyopathy and abdominal mass [6,7]. In 2009, Choi et al. [6] reported a case in which a severe catecholamine-induced cardiomyopathy barely responded to high vasopressin and epinephrine. Therefore, to evaluate the possibilities of such cardiac abnormalities, an echocardiogram must also be performed.
PHEO/PGL are associated with genetic syndromes such as neurofibromatosis 1, von Hippel-Lindau (VHL) disease, multiple endocrine neoplasia type 1 (MEN 1) and type 2 (MEN 2) [5,8]. Abdominal PGLs are also associated with succinate dehydrogenase subunit D (SDHD) B mutation [5]. Thus, medical information obtained through preoperative genetic counseling can aid in diagnosing PHEO/PGL.
In the case described here, the patient was admitted to the hospital for surgical treatment of an incidental duodenal mass. The case was uneventful not only prior to the surgery, but also during endotracheal intubation and the surgical incision. However, direct manipulation of the tumor led to hypertensive crisis in this patient. The patient's vital signs returned to normal after discontinuation of tumor manipulation. This finding is believed to be due to the sudden excessive secretion of catecholamines from the tumor (extra-adrenal or retroperitoneal PGL), which was misdiagnosed as GIST. GISTs are large-sized tumors (> 5 cm), arising from the stomach, with heterogeneous enhancement (84%) and central necrosis (37%) [9]. Large-sized PGLs adjacent to the stomach, accompanied by no specific signs of catecholamine secretion, are therefore often misdiagnosed as tumors of neural or mesodermal origin, such as GIST or other soft tissue sarcomas. Furthermore, when there are neither symptoms of catecholamine excess nor histologic diagnosis, GIST has a similar radiographic appearance to PGL [10]. The genetic association of nonfamilial PGL, GIST, and pulmonary chondroma (Carney triad) has been described in a very small number of patients, but the underlying genetic factor has not yet been revealed [11]. A few reports exist on misdiagnosis of PGL. In these cases, what was initially perceived by physicians to be GIST turned out, after biopsy, to be PGL. In a case described in 2009, Lowenthal et al. [7] believed that a patient with anxiety symptoms and a perigastric mass had GIST. While undergoing a surgical resection, the patient experienced a hypertensive crisis during the initial tumor manipulation. From the pathologic examination, the patient was diagnosed with PGL. In another case, in 2007, Yoo et al. [12] discovered an incidental duodenal polypoid mass that they thought was an adenoma. The biopsy results from the wedge resection led to the diagnosis of PGL. There are reports of PGL occurring not only in the duodenum but also in the liver, urethra, cerebellopontine angle, cauda equina, and larynx, among other sites [13]. Therefore, the surgeon must be aware of the fact that one could encounter PGL in unexpected parts of the body during surgery. Imaging studies for localizing the tumor and staging the disease are important, and the conventional work-up with CT or MRI is recommended. Since 123I-metaiodobenzylguanidine (MIBG) shows higher specificity in confirming catecholamine-secreting adrenergic tissue, this is the method of choice for diagnosis. However, finding the correlation between the symptoms and laboratory values is considered the most important, even if PGL is seen on a CT.
In cases where PGL is suspected due to a sudden hypertensive crisis during surgery, one must decide whether to proceed with the surgical procedure or to stop the surgery and restart it after proper management of the crisis. In the case discussed here, the surgeon continued with the surgical procedure because the patient's vital signs were stable after the infusion of short-acting sodium nitroprusside and nicardipine hydrochloride. Sodium nitroprusside is effective in arteriolar dilatation and in suppressing the hypertensive response due to circulating catecholamines. There have been recent reports that nicardipine hydrochloride suppresses the secretion of norepinephrine in PHEO/PGL, and its use has therefore increased [14]. Surgical procedures involving an unexpected PGL without proper preoperative management, however, lead to an increase in morbidity and mortality. Elective surgical procedures should be canceled or delayed until the origin of the hypertensive episode can be clarified; furthermore, a differential diagnosis must be made and proper preoperative medication with ╬▒1-blockers must be administered to the patient. When proper preoperative management is done, perioperative mortality can be reduced from 45% to 0-3% [15]. Once the diagnosis of PGL has been confirmed, complete surgical resection of the tumor is the treatment of choice. To minimize the chances of cerebrovascular catastrophes and/or cardiovascular complications such as arrythmias, hemodynamic instability, and even hypertensive crisis during the perioperative period, at least 1-2 weeks of preoperative medical treatment with a nonselective ╬▒1-adrenergic blocker (phenoxybenzamine) or a selective post-synaptic ╬▒1-adrenergic blocker (doxazosin, prazosin, or terazocin) is recommended. After adequate ╬▒-blockers have been administered, ╬▓-blockers can be added to prevent catecholamine-induced arrhythmias or reflex tachycardia, which are the side effects of ╬▒-blockers.
Although the majority of PGL are benign tumors, they may also be malignant, especially in children. Approximately 12% of PGL are malignant [8]. Several imaging studies, as well as cytologic and histologic studies by biopsy, are useful in suggesting malignancy. The combination of large size, heterogeneous density, irregular margins, and distant metastasis increases the possibility of malignancy, but the presence of distant metastasis is the most important factor. Chromogranin A is a valuable tumor marker that correlates with size and malignancy, and is useful for detecting SDHB-related PGL, which is biochemically silent [8]. This marker may improve the sensitivity of diagnosis, and it can be used effectively to monitor for recurrence during long-term follow-up. Although the initial therapy recommended for PGL is surgical resection, in cases of metastatic or unresectable disease, 123I-MIBG therapy is the standard alternative therapy option. Careful follow-up is needed in these situations.
In conclusion, PGL is very difficult to diagnose during preoperative patient evaluation, as it has either no symptoms or atypical symptoms. Therefore, if acute hemodynamic instability, such as malignant hypertension and tachycardia, occurs during induction of anesthesia or surgical manipulation, particularly in the case of GIST surgery, one should suspect PGL. Being aware of the lesion and the potential for hypertensive crisis may prevent intraoperative cardiac disaster. In order to minimize the risk of PGL removal, anticipation by the anesthesiologist and guarded manipulation by the surgeon are essential.