Introduction
Recently, we reported that ╬║-opioid receptor (OR) activation by U50488H (trans-(┬▒)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide hydrochloride) during the early reperfusion phase reduced both myocardial infarction and stunning in isolated rat hearts [1]. Infarct size was reduced to more than 50% and cardiodynamic functional variables were well preserved compared to untreated control hearts.
A marked increase in intracellular Ca2+ content ([Ca2+]i) has also been reported in myocardial ischemia/reperfusion (I/R) injury [2,3]. Ca2+ overload is a major cause of myocardial cell damage and cardiac dysfunction in ischemic hearts, and Ca2+ overload evoked by reperfusion is associated with irreversible myocardial injury [4]. Cardiac L-type Ca2+ channels are the main venue for Ca2+ influx into cardiac cells, and the increase in[Ca2+]i likely results in the depressed ability of I/R injured hearts to generate contractile force [5,6]. Therefore, inhibition of calcium channels has been proposed as an effective strategy against myocardial I/R injury [7,8]. Interestingly, U50488H is known to have an OR stimulation action as well as a non-opioid action that involves L-type Ca2+ channel blockade, which is not antagonized by OR antagonism [9]. In this regard, one cannot rule out the role of Ca2+ channels in U50488H-induced cardioprotection.
In the present study, we used a Ca2+ channel activator in an isolated rat heart model to investigate if modulation of Ca2+ channels plays a role in terms of both infarct size limitation and functional recovery in U50488H-mediated cardioprotection.
Materials and Methods
Langendorff isolated heart perfusion preparation
The experimental procedures and protocols used in this study were reviewed and approved by our Institutional Animal Care and Use Committee. Male Sprague-Dawley rats, weighing 280-330 gm (obtained from Korea Taconic, Republic of Korea), were used for the experiments. They received 100 mg/kg of pentobarbital sodium (Entobar®, Hanlim Pharmacy, Yongin-si, Korea) and 300 IU of heparin intraperitoneally. Hearts were isolated and perfused with modified Krebs-Henseleit (KH) solution containing 118.5 NaCl, 4.7 KCl, 1.2 MgSO4, 1.8 CaCl2, 24.8 NaHCO3, 1.2 KH2PO4, and 10 glucose (in mM) as described in our previous reports [1,10].
Ischemia and reperfusion induction
Regional ischemia and reperfusion were induced as described in our previous report [10]. In brief, the proximal length of the left coronary artery (LCA) was localized followed by passage of a 6-0 polypropylene suture around the major trunk of the LCA or its prominent branches. The ends of the thread were passed through a small piece of PE 50 plastic tube to form a snare. Regional ischemia was induced by pulling the snare and confirmed by regional cyanosis, a substantial decrease in left ventricular developed pressure (LVDP), or a fall of coronary flow (CF). Reperfusion was induced by releasing the ends of the suture.
Assessment of cardiac function
In isolated hearts, a latex balloon was inserted into the left ventricle through the left atrial appendage. Balloon volume was adjusted with KH buffer to 5-10 mmHg of the left ventricular end-diastolic pressure (LVEDP) at the beginning of the experiment. CF was measured by timed collection of the perfusate dripping from the right heart into a graduated cylinder.
Hemodynamic data, including heart rate (HR), left ventricular systolic pressure (LVSP), and LVEDP, were continuously recorded with the MP150 system (BIOPAC Systems Inc., CA). LVDP and rate-pressure product (RPP) were calculated as follows LVDP = LVSP - LVEDP and RPP = LVDP ├Ś HR. The maximum and minimum of the first derivative of left ventricular pressure (+dP/dtmax and -dP/dtmin) were analyzed with the Acqknowledge software, version 3.9.0.
Experimental protocol
All hearts were allowed to stabilize for at least 20 min after hanging on the hearts to Langendorff apparatus and were subjected to 30 min of regional ischemia and 2 h of reperfusion. Each group consisted of at least 7 hearts. Isolated rat hearts were assigned randomly to one of the following six groups:
CON (n = 9); control, no intervention either before or after LCA occlusion,
U50 (n = 8); 1 ┬ĄM of the standard selective ╬║-OR agonist U50488H,
U50+BNI (n = 7); 1 ┬ĄM U50488H combined with 5 ┬ĄM of the standard ╬║-OR antagonist nor-binaltorphimine (nor-BNI),
U50+BAY (n = 8); 1 ┬ĄM U50488H combined with 10 ┬ĄM of the L-type Ca2+ channel activator BAY K 8644 (1,4-dihydro-2,6-dimethyl-5-nitro-4-(-2-(trifluoromethyl)-phenyl)-3-pyrinecarboxylic acid methyl ester),
BNI (n = 7); nor-BNI alone, and
BAY (n = 7); BAY K 8644 alone.
U50488H, nor-BNI, and BAY K 8644 were purchased from Tocris Bioscience (Ellisville, MO, USA). The compounds were padministered over a period of 5 min before reperfusion, then again for 30 min after reperfusion. U50488H and nor-BNI were dissolved in distilled water and BAY K 8644 was dissolved in dimethyl sulfoxide. The concentrations of all chemicals were based on previous studies using isolated working rat hearts [1,11,12].
Determination of area at risk and infarct size
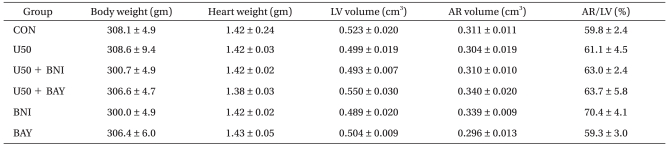
At the end of each experiment, the area at risk (AR) and area of necrosis (AN) were measured as described previously [10]. Briefly, the LCA perfusion circuit was reoccluded, and diluted fluorescent polymer microspheres (Duke Scientific Corp., Palo Alto, MA, USA) were infused to demarcate the AR. The hearts were cut into 2-mm thick transverse slices using a rat heart slice matrix (Zivic Instruments, Pittsburgh, PA, USA). The slices were incubated in 1% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich Chemical, St. Louis, MO, USA) in sodium phosphate buffer (pH = 7.4) at 37Ōäā for 20 min and subsequently immersed in 10% formalin to enhance the contrast. The myocardial AR was identified by illuminating the slices with UV light. The infarcted (unstained with TTC) and risk zone regions (no fluorescent area) were traced on a clear acetate transparent sheet and quantified with UTHSCSA Image Tool, version 3.0 (Department of Dental Diagnostic Science at The University of Texas Health Science Center, San Antonio, Texas, USA). The areas were converted into volumes by multiplying them by slice thickness. The AN volume was expressed as a percentage of the AR volume. All measurements were performed in a blinded fashion. There were no significant group differences with respect to body weight, heart weight, and LV volume (Table 1).
Statistical analysis
Data are presented as means ┬▒ SEM. Data analysis was performed with a personal computer statistical software package (SPSS for Windows, Release 12.0 SPSS Inc, Chicago, IL, USA). Data were analyzed using one-way analysis of variance (ANOVA) with Bonferroni post-hoc testing. Differences were considered to be statistically significant when P values were < 0.05.
Results
Forty-six rat hearts were used for this experiment. All hearts were perfused within 30 sec after excision. Ventricular fibrillation (VF) occurred in 15 of 46 hearts (3/9 in CON, 2/8 in U50, 3/7 in U50 + BNI, 3/8 in U50+BAY, 2/7 in BNI, and 2/7 in BAY) during early reperfusion. Hearts experiencing VF usually revert spontaneously to sinus rhythm. VF that lasted more than 30 sec was treated with finger flick cardioversion until a perfusing rhythm was obtained. Statistical analysis was not performed for the occurrence of VF because of the small sample size in each group.
Determination of infarct size
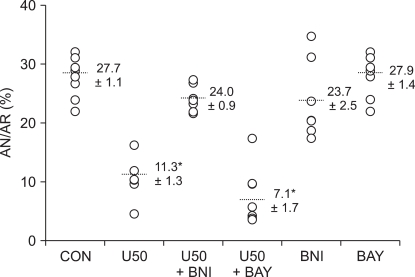
Risk volume averaged 0.296 cm3 to 0.340 cm3 with no statistically significant differences among groups (P > 0.05). The AR/LV ranged from 59.3% to 70.4% with no significant differences among all groups (P > 0.05), suggesting that the changes in infarct size observed between various groups in our experiments were not related to the degree of ischemic area.
As shown in Fig. 1, the area of infarct in the control hearts was 27.7 ┬▒ 1.1% of the AR, which is in agreement with our recently reported infarct measurement study [1,10]. U50488H treatment targeting reperfusion significantly reduced infarct size (11.3 ┬▒ 1.3%) compared to untreated control hearts (P < 0.001). The U50488H-induced infarct limitation effect was completely attenuated by the selective ╬║-OR antagonist nor-BNI (24.0 ┬▒ 0.9%, P < 0.001 vs. U50) but not by the L-type Ca2+ channel activator BAY K 8644 (7.1 ┬▒ 1.7%, P < 0.001 vs. CON). In the rats that received only nor-BNI or BAY K 8644, neither nor-BNI (23.7 ┬▒ 2.5%) nor BAY K 8644 (27.9 ┬▒ 1.4%) alone altered infarct size (P > 0.05 vs. CON).
Cardiac functional recovery
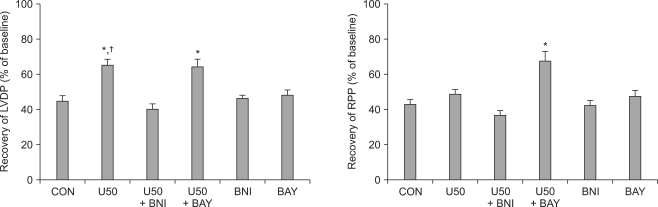
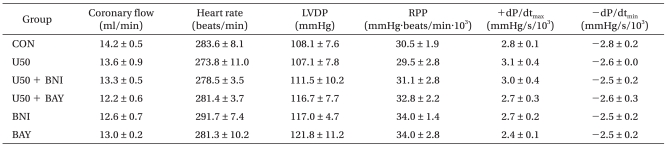
There were no significant baseline differences in CF or in hemodynamic indices, namely HR, LVDP, RPP, +dP/dtmax, and -dP/dtmin among the groups (Table 2). CF abruptly decreased after ischemia and diminished continuously during reperfusion without significant differences among groups (data not shown). After 2 h of reperfusion, the LVDP, RPP, +dP/dtmax, and -dP/dtmin in control hearts were 44.8 ┬▒ 3.6%, 42.9 ┬▒ 3.8%, 47.8 ┬▒ 3.3%, and 45.0 ┬▒ 1.9% of the baseline levels, respectively, and these results are in agreement with a previous report in isolated rat hearts [1]. HR was significantly decreased in U50488H-treated hearts (P < 0.01 vs. CON, Fig. 2). Both BNI and BAY K 8644 completely abrogated the decrease in HR in U50488H-treated hearts (P < 0.05 vs. U50, Fig. 2).
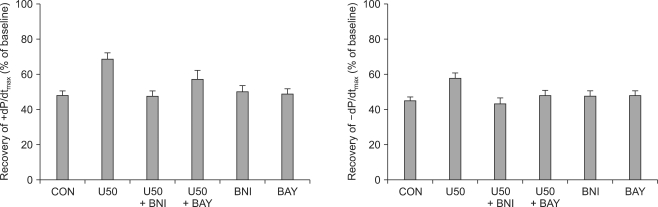
As shown in Fig. 3, U50488H significantly increased LVDP after reperfusion (65.3 ┬▒ 4.8%, P < 0.05 vs. CON). The increase in LVDP by U50488H was abrogated by nor-BNI (40.5 ┬▒ 4.5%, P < 0.01 vs. U50) but not by BAY K 8644 (64.3 ┬▒ 5.6% P > 0.05 vs. U50). There was no significant change in RPP between U50488H-treated hearts (49.1 ┬▒ 3.6%) and untreated control hearts. Administration of BAY K 8644 in U50488H-treated hearts (68.1 ┬▒ 6.4%) significantly increased RPP compared to both control and U50488H-treated hearts (P < 0.01). +dP/dtmax and -dP/dtmin exhibited a tendency to increase in U50488H-treated hearts but there were no significant differences compared to control hearts (P > 0.05, Fig. 4). When control hearts were compared to hearts first treated with U50488H then with either BNI or BAY K 8644, there were no significant differences in cardiodynamic variables.
Discussion
Because U50488H inhibits L-type Ca2+ currents in guinea pig ventricular myocytes without OR binding involvement [9] and U50488H increases [Ca2+]i in isolated rat ventricular myocytes, an effect that is antagonized by ╬║-OR antagonists [13], there is a controversy about the role of Ca2+ channels in U50488H-induced cardioprotection.
In the present study, treatment with the ╬║-OR agonist U50488H at reperfusion significantly decreased myocardial infarction and stunning in isolated rat hearts; these results are consistent with our recent previous report [1]. U50488H-mediated infarct size limitation was totally abrogated by the ╬║-OR antagonist nor-BNI, but not by the L-type Ca2+ channel activator BAY K 8644, implying that the infarct size limitation effect by U50488H at reperfusion is solely caused by OR activation and not by Ca2+ channel inhibition. In addition, U50488H significantly improved myocardial contractility, as reflected by LVDP, after reperfusion. LVDP recovered to 65.3% of the baseline level (44.8% in U50488H-untreated control hearts). The increase in LVDP was completely blocked by nor-BNI, suggesting that the recovery of LVDP associated with U50488H treatment was induced by ╬║-OR activation. However, BAY K 8644 could not attenuate U50488H-mediated recovery of LVDP, which argues against L-type Ca2+ channel involvement. Taken together, our data strongly suggest that cardioprotection by U50488H is mediated by opiodergic regulation but not by Ca2+ channel modulation.
U50488H treatment targeting reperfusion significantly decreased HR. Notably, this effect was completely blocked by both nor-BNI and BAY K 8644. It has been demonstrated that BAY K 8644 treatment itself may increase or not change HR [14,15]. Though our concentration (10 ┬ĄM) of BAY K 8644 by itself did not alter HR compared to control hearts, this concentration of BAY K 8644 completely attenuated the decrease in HR by U50488H. This result strongly suggests that the decrease in HR by U50488H is modulated by both OR activation and Ca2+ channel inhibition. The RPP (product of LVDP and HR) in U50488H-treated hearts was not statistically different from that in U50488H-untreated control hearts, although U50488H significantly increased LVDP after reperfusion. This result might be secondary to the combination of the decrease in HR and the increase in LVDP induced by U50488H.
It is well accepted that Ca2+ ions are major regulators of cardiac function. Hypoxia and ischemia produce depression of myocardial contractile function and alterations in Ca2+ homeostasis and reoxygenation or reperfusion can also lead to paradoxical augmentation of injury [16]. It appears as if [Ca2+]i overload is a recognized common pathway that underlies myocardial I/R injury [17]. The L-type Ca2+ channel, a type of voltage-dependent Ca2+ channel, is considered the most significant Ca2+ entry portal into cardiac myocytes. The small amount of Ca2+ entering the cytosol through this channel triggers the release of additional Ca2+ from the sarcoplasmic reticulum. Accordingly, it has been proposed that lowering sarcolemmal Ca2+ content may be a mechanism underlying cardioprotective and anti-arrhythmic features [18,19] and that inhibition of Ca2+ channels may protect I/R injured myocardium by retardation of an early rise in [Ca2+]i [20]. Indeed, there are several lines of evidence that Ca2+ channel antagonists effectively treat myocardial I/R injury [8,21,22]. However, in our study, U50488H reduced myocardial infarction and stunning via OR stimulation but not by Ca2+ channel modulation, even though there is evidence that U50488H also inhibits L-type Ca2+ channels in ventricular myocytes [9].
In conclusion, the ╬║-OR agonist U50488H significantly limits myocardial infarction and stunning in I/R-injured isolated rat hearts. The infarct size limitation by U50488H is mediated by OR stimulation but not by Ca2+ channel modulation. In addition, the improved contractility by U50488H is also induced by OR activation and not by Ca2+ channel inhibition. In contrast, the decrease in HR by U5048H is mediated by both OR activation and Ca2+ channel inhibition.